Stille reaction
[6][2][7][8][9][10][11][12][13][14][15][excessive citations] The first example of a palladium catalyzed coupling of aryl halides with organotin reagents was reported by Colin Eaborn in 1976.
This process was expanded to the coupling of acyl chlorides with alkyl-tin reagents in 1977 by Toshihiko Migita, yielding 53% to 87% ketone product.
John Kenneth Stille subsequently reported the coupling of a variety of alkyl tin reagents in 1978 with numerous aryl and acyl halides under mild reaction conditions with much better yields (76%–99%).
[18][20] Stille continued his work in the 1980s on the synthesis of a multitude of ketones using this broad and mild process and elucidated a mechanism for this transformation.
Like other palladium-catalyzed coupling reactions, the active palladium catalyst is believed to be a 14-electron Pd(0) complex, which can be generated in a variety of ways.
First, a bulky ligand set is usually used in these processes, such as phosphines, and it is highly unfavorable for them to adopt a cis orientation relative to each other, resulting in isomerization to the more favorable trans product.
[29][30] Since halides or pseudohalides are significantly more electronegative, their bonding with palladium will be highly polarized, with most of the electron density on the X group, making them low trans effect ligands.
[24][28][30] The transmetallation of the trans intermediate from the oxidative addition step is believed to proceed via a variety of mechanisms depending on the substrates and conditions.
[31] First, when the organostannane initially adds to the trans metal complex, the X group can coordinate to the tin, in addition to the palladium, producing a cyclic transition state.
Another commonly seen mechanism involves the same initial addition of the organostannane to the trans palladium complex as seen above; however, in this case, the X group does not coordinate to the tin, producing an open transition state.
When the solvent detaches, to form a 14-electron trivalent intermediate, the organostannane can add to the palladium, undergoing an open or cyclic type process as above.
This reaction occurs in two steps: first, the reductive elimination is followed by coordination of the newly formed sigma bond between R1 and R2 to the metal, with ultimate dissociation yielding the coupled product.
The chloride ion is believed to either displace the X group on the palladium making the catalyst more active for transmetalation or by coordination to the Pd(0) adduct to accelerate the oxidative addition.
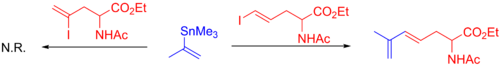
A variety of alkenes may be used, and these include both α- and β-halo-α,β unsaturated ketones, esters, and sulfoxides (which normally need a copper (I) additive to proceed), and more (see example below).
[10] Acyl chlorides are also used as coupling partners and can be used with a large range of organostannane, even alkyl-tin reagents, to produce ketones (see example below).
An alternative developed to this process is the Stille-carbonylative cross-coupling reaction, which introduces the carbonyl group via carbon monoxide insertion.
While commonly employed, allylic halides proceed via an η3 transition state, allowing for coupling with the organostannane at either the α or γ position, occurring predominantly at the least substituted carbon (see example below).
Larry Overman's 19-step enantioselective total synthesis of quadrigemine C involves a double Stille cross metathesis reaction.
Panek's 32 step enantioselective total synthesis of ansamycin antibiotic (+)-mycotrienol makes use of a late stage tandem Stille type macrocycle coupling.
[6][55] Stephen F. Martin and coworkers' 21 step enantioselective total synthesis of the manzamine antitumor alkaloid Ircinal A makes use of a tandem one-pot Stille/Diels-Alder reaction.
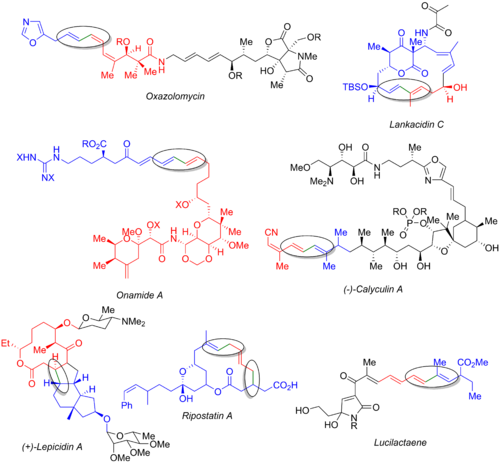
[6][56] Numerous other total syntheses utilize the Stille reaction, including those of oxazolomycin,[57] lankacidin C,[58] onamide A,[59] calyculin A,[60] lepicidin A,[61] ripostatin A,[62] and lucilactaene.
[6][63] The image below displays the final natural product, the organohalide (blue), the organostannane (red), and the bond being formed (green and circled).
From these examples, it is clear that the Stille reaction can be used both at the early stages of the synthesis (oxazolomycin and calyculin A), at the end of a convergent route (onamide A, lankacidin C, ripostatin A), or in the middle (lepicidin A and lucilactaene).
The synthesis of lucilactaene features a middle subunit, having a borane on one side and a stannane on the other, allowing for Stille reactionfollowed by a subsequent Suzuki coupling.
[64][65] In the realm of green chemistry a Stille reaction is reported taking place in a low melting and highly polar mixture of a sugar such as mannitol, a urea such as dimethylurea and a salt such as ammonium chloride[66] .
[67] The catalyst system is tris(dibenzylideneacetone)dipalladium(0) with triphenylarsine: A common alteration to the Stille coupling is the incorporation of a carbonyl group between R1 and R2, serving as an efficient method to form ketones.
This process is extremely similar to the initial exploration by Migita and Stille (see History) of coupling organostannane to acyl chlorides.
The CO can coordinate to the palladium catalyst (9) after initial oxidative addition, followed by CO insertion into the Pd-R1 bond (10), resulting in subsequent reductive elimination to the ketone (12).
[6][69] Louis Hegedus' 16-step racemic total synthesis of Jatraphone involved a Stille-carbonylative cross-coupling as its final step to form the 11-membered macrocycle.
[6] Jie Jack Lie et al. made use of the Stille-Kelly coupling in their synthesis of a variety of benzo[4,5]furopyridines ring systems.


























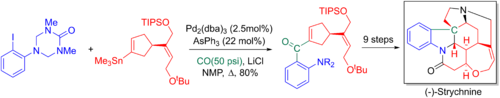



![Synthesis of benzo[4,5]furopyridines](http://upload.wikimedia.org/wikipedia/commons/thumb/2/25/Benzofuropyridines.png/500px-Benzofuropyridines.png)