Acute myeloid leukemia
[1] Symptoms may include feeling tired, shortness of breath, easy bruising and bleeding, and increased risk of infection.
[1] Risk factors include getting older, being male,[6] smoking, previous chemotherapy or radiation therapy, myelodysplastic syndrome, and exposure to the chemical benzene.
[1][3] The specific genetic mutations present within the cancer cells may guide therapy, as well as determine how long that person is likely to survive.
[2] Most signs and symptoms of AML are caused by the crowding out in bone marrow of space for normal blood cells to develop.
[8] A low red blood cell count (anemia) can cause fatigue, paleness, shortness of breath and palpitations.
[11] Other blood disorders, particularly myelodysplastic syndrome (MDS) and less commonly myeloproliferative neoplasms (MPN), can evolve into AML;[9] the exact risk depends on the type of MDS/MPN.
[14] Historically, survivors of the atomic bombings of Hiroshima and Nagasaki had an increased rate of AML,[15] as did radiologists exposed to high levels of X-rays prior to the adoption of modern radiation safety practices.
[11] For reasons that may relate to substance or radiation exposure, certain occupations have a higher rate of AML; particularly work in the nuclear power industry, electronics or computer manufacturing, fishing and animal slaughtering and processing.
In AML, though, a single myeloblast accumulates genetic changes which stop maturation, increase its proliferation, and protect it from programmed cell death (apoptosis).
[19] The chromosomal translocations encode abnormal fusion proteins, usually transcription factors whose altered properties may cause the "differentiation arrest".
[20] For example, in APL, the t(15;17) translocation produces a PML-RARA fusion protein which binds to the retinoic acid receptor element in the promoters of several myeloid-specific genes and inhibits myeloid differentiation.
[18] A sample of marrow or blood is typically also tested for chromosomal abnormalities by routine cytogenetics or fluorescent in situ hybridization.
[28][29] According to the WHO criteria, the diagnosis of AML is established by demonstrating involvement of more than 20% of the blood and/or bone marrow by leukemic myeloblasts, except in three forms of acute myeloid leukemia with recurrent genetic abnormalities: t(8;21), inv(16) or t(16;16), and acute promyelocytic leukemia with PML-RARA, in which the presence of the genetic abnormality is diagnostic irrespective of blast percent.
[30][31] The older French–American–British (FAB) classification, which is no longer widely used,[29] is a bit more stringent, requiring a blast percentage of at least 30% in bone marrow or peripheral blood for the diagnosis of AML.
Fluorescent in situ hybridization performed on blood or bone marrow is often used for this purpose, as it readily identifies the chromosomal translocation [t(15;17)(q22;q12);] that characterizes APL.
Each of the WHO categories contains numerous descriptive subcategories of interest to the hematopathologist and oncologist; however, most of the clinically significant information in the WHO schema is communicated via categorization into one of the subtypes listed below.
This classification, which is based on a combination of genetic and immunophenotypic markers and morphology, defined the subtypes of AML and related neoplasms as shown below.
Classification is done by examining the appearance of the malignant cells with light microscopy and/or by using cytogenetics to characterize any underlying chromosomal abnormalities.
[60] Acute promyelocytic leukemia is treated with all-trans-retinoic acid (ATRA) and either arsenic trioxide (ATO) monotherapy or an anthracycline.
[64] There is insufficient evidence to determine if prescribing ATRA in addition to chemotherapy to adults who have other subtypes of acute myeloid leukaemia is helpful.
[65] Even after complete remission is achieved, leukemic cells likely remain in numbers too small to be detected with current diagnostic techniques.
This generally involves cytarabine, with the doses administered being higher in younger patients, who are less likely to develop toxicity related to this treatment.
The FDA has approved certain epigenetic modifying drugs like ivosidenib and enasidenib, which are used in patients that can no longer receive intensive induction chemotherapy; specifically, they are involved in the therapy of IDH1 and IDH2 mutations.
Two other mutations – NPM1 and biallelic CEBPA are associated with improved outcomes, especially in people with normal cytogenetics and are used in current risk stratification algorithms.
These are prevalent, and potentially clinically relevant because of the availability of tyrosine kinase inhibitors, such as imatinib and sunitinib that can block the activity of c-KIT pharmacologically.
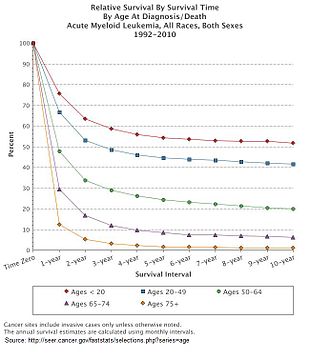
[11] As with most forms of cancer, performance status (i.e. the general physical condition and activity level of the person) plays a major role in prognosis as well.
[91] The first published description of a case of leukemia in medical literature dates to 1827 when French physician Alfred-Armand-Louis-Marie Velpeau described a 63-year-old florist who developed an illness characterized by fever, weakness, urinary stones, and substantial enlargement of the liver and spleen.
[92] In 1845, a series of people who died with enlarged spleens and changes in the "colors and consistencies of their blood" was reported by the Edinburgh-based pathologist J.H.
[95] The term "myeloid" was coined by Franz Ernst Christian Neumann in 1869, as he was the first to recognize white blood cells were made in the bone marrow (Greek: μυєλός, myelos, lit.
[96] Finally, in 1900, the myeloblast, which is the malignant cell in AML, was characterized by Otto Naegeli, who divided the leukemias into myeloid and lymphocytic.