Azomethine ylide
Azomethine ylides thus have high utility in total synthesis, and formation of chiral ligands and pharmaceuticals.
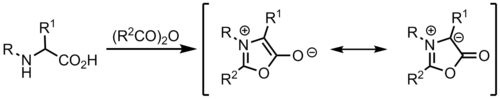
The resonance structures below show the 1,3-dipole contribution, in which the two carbon atoms adjacent to the nitrogen have a negative or positive charge.
[4][5] In accordance with the Woodward–Hoffmann rules, the thermal four-electron ring opening proceeds via a conrotatory process, whereas the photochemical reaction is disrotatory.
If the amine contains an electron-withdrawing group on the alpha carbon, such as an ester, the deprotonation occurs readily.
The reaction is generally viewed as concerted, in which the two carbon-carbon bonds are being formed at the same time, but asynchronously.
1,3-Dipolar cycloaddition reactions of azomethine ylides commonly use alkenes or alkynes as dipolarophiles, to form pyrrolidines or pyrrolines, respectively.
[14] When the dipole and dipolarophile are part of the same molecule, an intramolecular cyclization reaction can lead to a polycyclic product of considerable complexity.
[15][16] Enantioselective cycloaddition of azomethine ylides using chiral catalysts was first described in a seminal work by Allway and Grigg in 1991.
The method described by Gong, et al. leads to an unexpected regiochemical outcome that does not follow electronic effects.
The sterics and geometry of the reacting phenyl ring play a major role in the success of the reaction.
[21] A cycloaddition of an azomethine ylide with an unactivated alkene was used in total synthesis of martinellic acid.
The ylide then reacts with an electron-deficient alkene on an indolinone, resulting in formation of a spirocyclic pyrrolidine and four contiguous stereocenters.
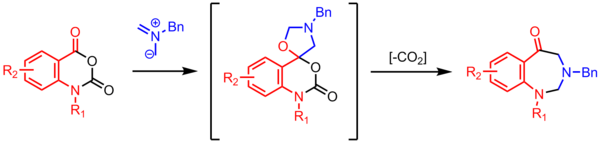
[23] Cyclization of an azomethine ylide with a carbonyl affords a spirocyclic oxazolidine, which loses CO2 to form a seven-membered ring.