BRAF (gene)
This protein plays a role in regulating the MAP kinase/ERKs signaling pathway, which affects cell division, differentiation, and secretion.
Residues 234–280 comprise a phorbol ester/DAG-binding zinc finger motif that participates in B-Raf membrane docking after Ras-binding.
[17][19] Residues 574–581 compose a section of the kinase domain responsible for supporting the transfer of the γ-phosphate of ATP to B-Raf's protein substrate.
In particular, D576 acts as a proton acceptor to activate the nucleophilic hydroxyl oxygen on substrate serine or threonine residues, allowing the phosphate transfer reaction to occur mediated by base-catalysis.
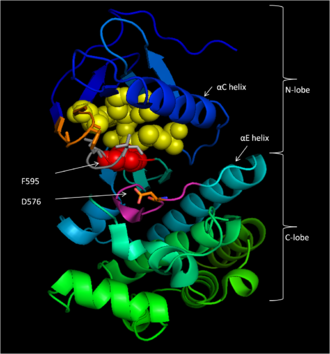
In the inactive state, F595 occupies the nucleotide-binding pocket, prohibiting ATP from entering and decreasing the likelihood of enzyme catalysis.
[15] Once B-Raf is activated, a conserved protein kinase catalytic core phosphorylates protein substrates by promoting the nucleophilic attack of the activated substrate serine or threonine hydroxyl oxygen atom on the γ-phosphate group of ATP through bimolecular nucleophilic substitution.
The DFG motif changes conformation with the activation loop, causing F595 to move out of the adenine nucleotide binding site and into a hydrophobic pocket bordered by the αC and αE helices.
[17] B-Raf binds ATP by anchoring the adenine nucleotide in a nonpolar pocket (yellow, Figure 1) and orienting the molecule through hydrogen-bonding and electrostatic interactions with phosphate groups.
K578 neutralizes the negative charge on the γ-phosphate group of ATP so that the activated ser/thr substrate residue will not experience as much electron-electron repulsion when attacking the phosphate.
[18][19][20] BAY43-9006 (Sorafenib, Nexavar) is a V600E mutant B-Raf and C-Raf inhibitor approved by the FDA for the treatment of primary liver and kidney cancer.
The hydrophobic interactions with catalytic loop F583 and DFG motif F595 stabilize the inactive conformation of these structures, decreasing the likelihood of enzyme activation.
Finally, polar interactions of BAY43-9006 with the kinase domain continue this trend of increasing enzyme affinity for the inhibitor and stabilizing DFG residues in the inactive conformation.
[18] The trifluoromethyl phenyl moiety cements the thermodynamic favorability of the inactive conformation when the kinase domain is bound to BAY43-9006 by sterically blocking the hydrophobic pocket between the αC and αE helices that the DFG motif and activation loop would inhabit upon shifting to their locations in the active conformation of the protein.
Ketone linker hydrogen bonding to water and difluoro-phenyl fit in a second hydrophobic pocket (A481, V482, K483, V471, I527, T529, L514, and F583) contribute to the exceptionally high binding affinity overall.
Inherited mutations in this gene cause cardiofaciocutaneous syndrome, a disease characterized by heart defects, mental retardation and a distinctive facial appearance.
[25] Mutations in this gene have been found in cancers, including non-Hodgkin lymphoma, colorectal cancer, malignant melanoma, papillary thyroid carcinoma, non-small-cell lung carcinoma, adenocarcinoma of the lung, brain tumors including glioblastoma and pleomorphic xanthoastrocytoma as well as inflammatory diseases like Erdheim–Chester disease.
[10] The V600E mutation of the BRAF gene has been associated with hairy cell leukemia in numerous studies and has been suggested for use in screening for Lynch syndrome to reduce the number of patients undergoing unnecessary MLH1 sequencing.
This leads to valine (V) being substituted for by glutamate (E) at codon 600 (now referred to as V600E) in the activation segment that has been found in human cancers.
[38] High frequency of BRAF V600E mutations have been detected in ameloblastoma, a benign but locally infiltrative odontogenic neoplasm.
Replacing the medium-sized hydrophobic Val side chain with a larger and charged residue as found in human cancer(Glu, Asp, Lys, or Arg) would be expected to destabilize the interactions that maintain the DFG motif in an inactive conformation, so flipping the activation segment into the active position.
As mentioned above, some pharmaceutical firms are developing specific inhibitors of mutated B-raf protein for anticancer use because BRAF is a well-understood, high yield target.
[19][42] Vemurafenib (RG7204 or PLX4032) was licensed by the US Food and Drug Administration as Zelboraf for the treatment of metastatic melanoma in August 2011 based on Phase III clinical data.
[44] While the mechanisms of this resistance are still disputed, some hypotheses include the overexpression of B-Raf to compensate for high concentrations of Vemurafenib[44] and upstream upregulation of growth signaling.
[46] BRAF (gene) has been shown to interact with: This article incorporates public domain material from Dictionary of Cancer Terms.