Congenital adrenal hyperplasia
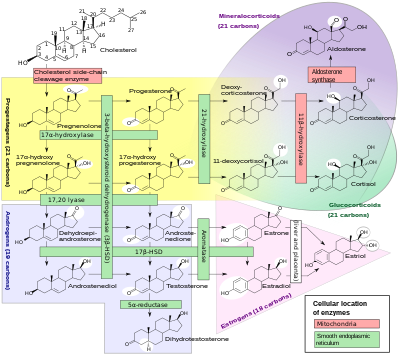
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterized by impaired cortisol synthesis.
The clinical presentation of each form is different and depends to a large extent on the underlying enzyme defect, its precursor retention, and deficient products.
[13] In 75% of cases of severe enzyme deficiency, insufficient aldosterone production can lead to salt wasting, failure to thrive, and potentially fatal hypovolemia and shock.
[2] The main feature of CAH in newborn females is the abnormal development of the external genitalia, which has varying degrees of virilization.
[14] The nonclassic form may be noticed in late childhood and may lead to signs of hyperandrogenism such as accelerated growth, acne, hirsutism, premature pubarche, menstrual irregularities,[14] and secondary polycystic ovary syndrome.
Milder degrees of inefficiency are usually associated with excessive or deficient sex hormone effects in childhood or adolescence, while the mildest forms of CAH interfere with ovulation and fertility in adults.
[citation needed] Female infants with classic CAH have ambiguous genitalia due to exposure to high concentrations of androgens in utero.
[medical citation needed] Genetic analysis can be helpful to confirm a diagnosis of CAH, but it is not necessary if classic clinical and laboratory findings are present.
Many neonatal screening programs have specific reference ranges by weight and gestational age because high levels may be seen in premature infants without CAH.)
Since the 1960s, most endocrinologists have referred to the forms of CAH by the traditional names in the left column, which generally correspond to the deficient enzyme activity.
[non-primary source needed] In 2020, Wael AbdAlmageed from USC Information Sciences Institute and Mimi Kim from USC Keck School Of Medicine led a joint study in which they used deep learning technology to analyze the facial morphology and features of CAH patients compared to control.
The findings suggest that facial morphologic features, as analyzed by deep neural network techniques, can be used as a phenotypic biomarker to predict CAH.
[citation needed] Since the clinical manifestations of each form of CAH are unique and depend to a large extent on the underlying enzyme defects, their precursor retention and defective products, the therapeutic goal of CAH is to replenish insufficient adrenal hormones and suppress excess of precursors.
In the United States, congenital adrenal hyperplasia in its classic form is particularly common in Native Americans and Yupik Inuit (incidence 1⁄280).
These were somewhat loose and resembled labia majora.De Crecchio then described the internal organs, which included a normal vagina, uterus, fallopian tubes, and ovaries.
I was determined to get as complete a story as possible, determined to get at the base of the facts and to avoid undue exaggeration which was rampant in the conversation of many of the people present at the time of the dissection.He interviewed many people and satisfied himself that Joseph Marzo "conducted himself within the sexual area exclusively as a male", even to the point of contracting the "French disease" (syphilis) on two occasions.
[36] This account was translated by Alfred Bongiovanni from De Crecchio ("Sopra un caso di apparenzi virili in una donna".
[citation needed] The association of excessive sex steroid effects with diseases of the adrenal cortex have been recognized for over a century.
Congenital adrenal hyperplasia, which also dates to the first half of the century, has become the preferred term to reduce ambiguity and to emphasize the underlying pathophysiology of the disorders.
[citation needed] Much modern understanding and treatment of CAH comes from research conducted at Johns Hopkins Medical School in Baltimore in the middle of the 20th century.
Lawson Wilkins, "founder" of pediatric endocrinology, worked out the apparently paradoxical pathophysiology: that hyperplasia and overproduction of adrenal androgens resulted from impaired capacity for making cortisol.
After application of karyotyping to CAH and other intersex disorders in the 1950s, John Money, JL Hampson, and JG Hampson persuaded both the scientific community and the public [citation needed] that sex assignment should not be based on any single biological criterion, and gender identity was largely learned and has no simple relationship with chromosomes or hormones.
The last decade, though, has seen a number of new developments, discussed more extensively in congenital adrenal hyperplasia due to 21-hydroxylase deficiency:[citation needed] Notable people with CAH include: