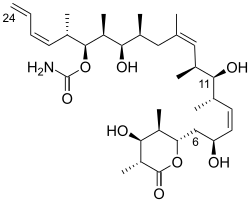
Discodermolide
(+)-discodermolide was isolated by Gunasekera and his co-workers at the Harbor Branch Oceanographic Institute from the deep-sea sponge Discodermia dissoluta in 1990.
(+)-discodermolide also shows some unique characters, including a linear backbone structure, immunosuppressive properties both in vitro and in vivo,[2][3] potent induction of an accelerated senescence phenotype,[4] and synergistic antiproliferative activity in combination with paclitaxel.
A large number of efforts toward the total synthesis of (+)-discodermolide were directed by its interesting biological activities and extreme scarcity of natural sources (0.002% w/w from frozen marine sponge).
[6] Discodermolide was first isolated in 1990 from the Caribbean marine sponge Discodermia dissoluta by chemist Dr. Sarath Gunasekera and biologist Dr. Ross Longley, scientists at the Harbor Branch Oceanographic Institution.
In both human peripheral blood leukocytes and murine splenocytes, (+)-discodermolide was found to suppress the two-way mixed lymphocyte reaction.
[12] It served as the “railroad track” upon which actin, tubulin, mitochondria, neurotransmitter-related enzymes and vesicles carrying messenger proteins are delivered.
In transgenic mouse model for human tauopathy, (+)-discodermolide stabilizes microtubules and restores fast axonal transport in cells, offsetting the loss of function caused by aggregation of tau protein.
Discodermolide competes with paclitaxel[13] for microtubule binding, but with higher affinity and is also effective in paclitaxel- and in epothilone-resistant cancer cells.
[14] An intense effort has been made towards the total synthesis of (+)-discodermolide in order to meet the growing interest of studying its clinical profile.
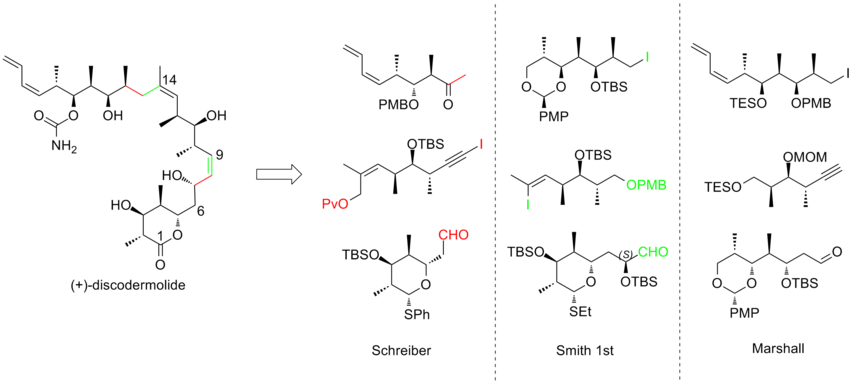
All of the total syntheses approaches started with the construction of three major fragments of roughly equivalent complexity, each of which contains the methyl-hydroxyl-methyl triad of contiguous centers that matches the stereogenicity of discodermolide target.
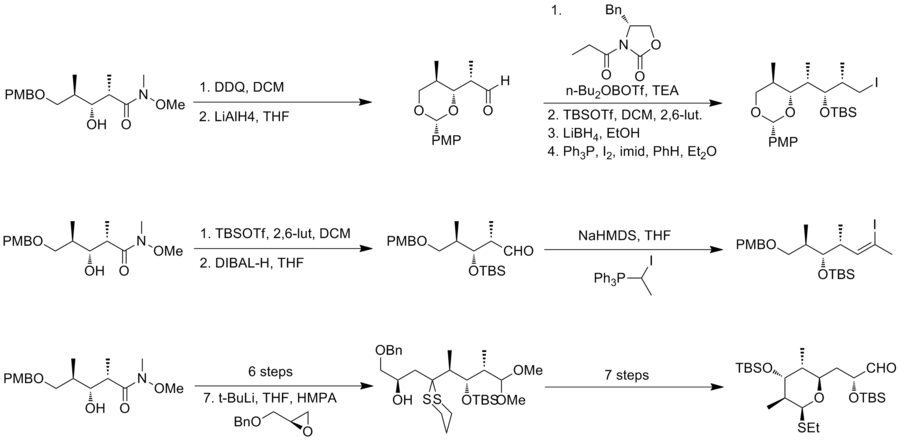
Here are three examples of the retrosynthetic analyses of (+)-discodermolide: In 1993, Schreiber and his co-workers[7] reported the first total synthesis of the unnatural antipode (-)-discodermolide and determined the absolute stereochemistry of the natural product.
The Schreiber team recognized three fragments of roughly equal complexity that are separated by olefinic units in discodermolide.
However, the undesired isomer can be recycled to the desired epimer in three steps, including Swern oxidation and Corey's asymmetric reduction.
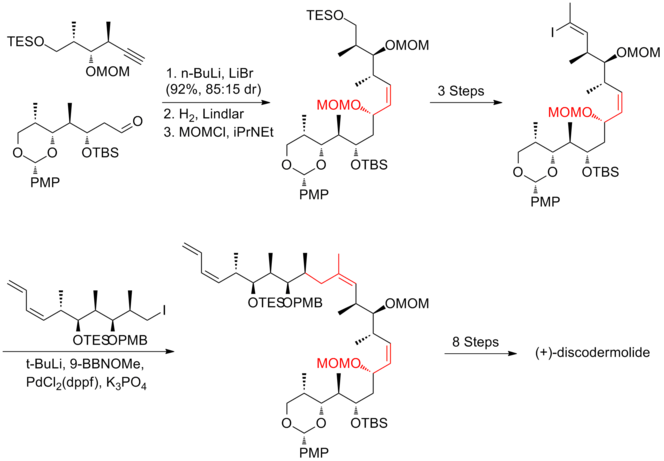
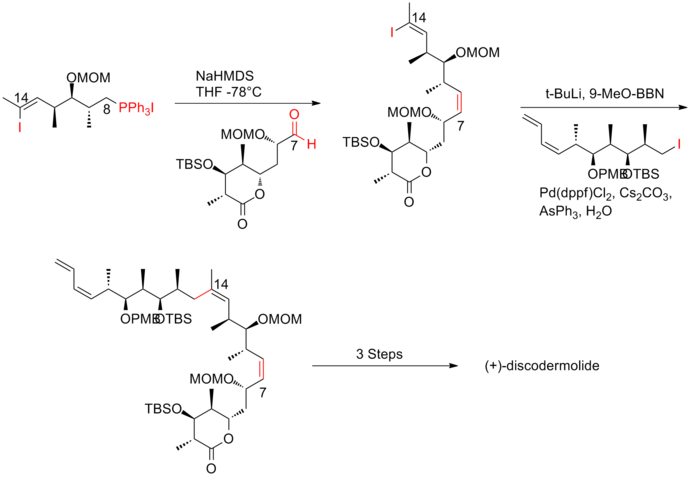
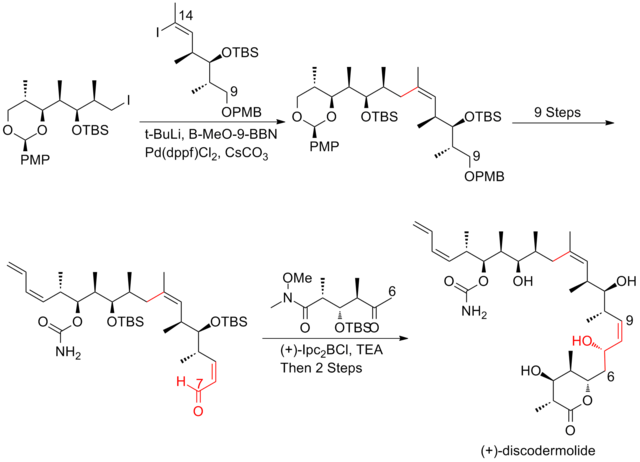
In Smith's strategy, all three fragments shared a common precursor, which was the product of a highly efficient 50g scale five-step conversion from 3-hydroxy-2-methylpropionate with 59% yield.
The titanium-mediated hetero-Diels–Alder reaction of aldehyde with the Danishefsky diene successfully produced the challenging Z-trisubstituted C(13)–C(14) olefin in allylic iodide fragment.
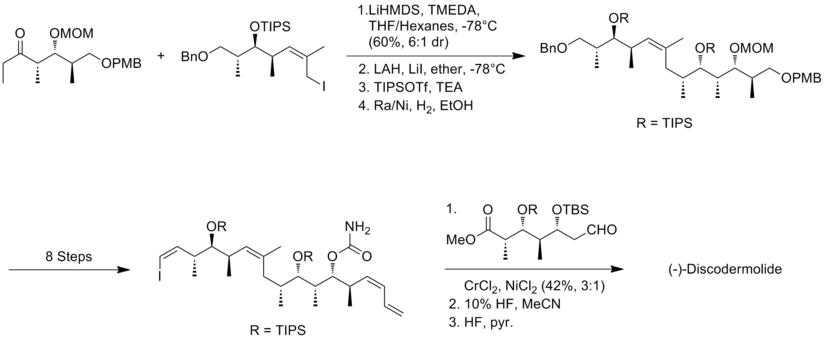
The novelty of the Marshall approach is that the three stereotriad subunits are assembled through addition of non-racemic allenylmetal reagents to (S)-3-silyloxy-2-methylpropanal to generate both syn/syn and syn/anti adducts.
The central feature for the synthesis of the alkyl iodide fragment was the treatment of aldehyde to allenyltributylstannane in the presence of BF3-etherate to get the syn/syn isomer.
The condensation of aldehyde with iodoethylidene triphenylphosphorane was the most challenging step, which produce 40% yield and an 85:15 inseparable mixture of (Z) and (E) isomers.
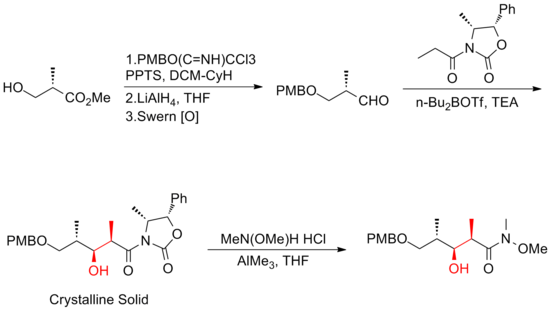
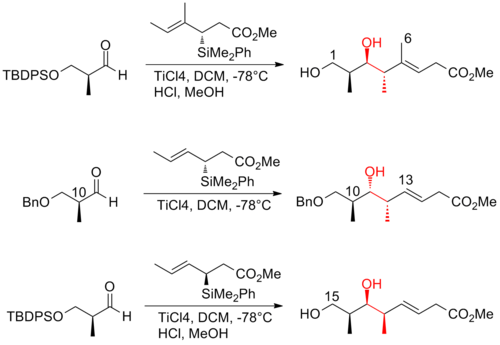
Evans and his co-workers[18] has developed a strategy that relies heavily on asymmetric aldol methodology for the production of the polypropionate backbone.
One of the major improvements was that no purification was required in the first four steps of the five-step sequence towards the common precursor as the intermediate, aldol adduct, is a crystalline solid.
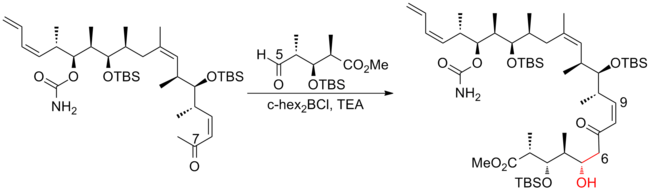
Paterson and his co-workers[21] at the University of Cambridge have developed a strategy which utilizes novel chelation-controlled and reagent-controlled aldol reactions with high selectivity for subunit connections.
Instead of a reagent-controlled aldol reaction in Paterson first-generation synthesis, a dicyclohexylboron-mediated anti-aldol was used to connect C(5)-C(6), which leads to a significant increase in diastereoselectivity from 4:1 to 92:8.
The stepwise method used in previous generations to incorporate the C(1)–C(8) subunit was replaced by a late-stage Still-Gennari olefination, which leads to a notable improvement in convergence.
This synthesis allows (+)-discodermolide to be evaluated as an in vivo chemotherapeutic agent for adult patients presenting with advanced solid malignancies in Phase I clinical trials.
In 2004, Panek and his co-workers[25] reported an approach which takes advantage of chiral crotylsilane-based C-C bond construction methodology to obtain the absolute stereochemistry of the three subunits of (+)-discodermolide.
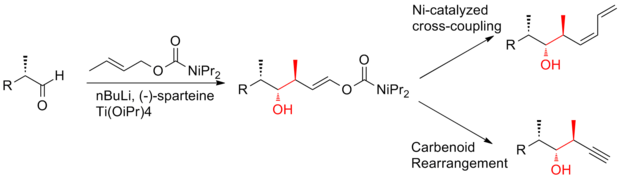
In 2008, Ardisson and his co-workers[26] reported a strategy that applies a crotyltitanation reaction repeatedly to yield homoallylic (Z)-O-ene-carbamate alcohols with excellent selectivity.
This crotyltitanation reaction not only efficiently produces the syn-anti methyl-hydroxy-methyl triads of (+)-discodermolide, but also yields products that can be easily converted to terminal (Z)-diene.
[27] Amos B. Smith's research group, in collaboration with Kosan Biosciences, has a preclinical drug development program ongoing.