FASTQ format
Both the sequence letter and quality score are each encoded with a single ASCII character for brevity.
It was originally developed at the Wellcome Trust Sanger Institute to bundle a FASTA formatted sequence and its quality data, but has become the de facto standard for storing the output of high-throughput sequencing instruments such as the Illumina Genome Analyzer.
Accounting for this makes parsing more complicated due to the choice of "@" and "+" as markers (as these characters can also occur in the quality string).
Early SRA loaders parsed these ids and stored their decomposed components internally.
NCBI stopped recording read names because they are frequently modified from the vendors' original format in order to associate some additional information meaningful to a particular processing pipeline, and this caused name format violations that resulted in a high number of rejected submissions.
The requirements for doing so have been dictated by users over several years, with the majority of early demand coming from the 1000 Genomes Project.
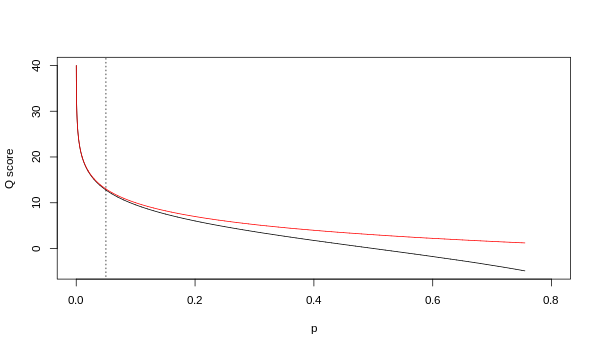
A quality value Q is an integer mapping of p (i.e., the probability that the corresponding base call is incorrect).
The first is the standard Sanger variant to assess reliability of a base call, otherwise known as Phred quality score: The Solexa pipeline (i.e., the software delivered with the Illumina Genome Analyzer) earlier used a different mapping, encoding the odds p/(1-p) instead of the probability p: Although both mappings are asymptotically identical at higher quality values, they differ at lower quality levels (i.e., approximately p > 0.05, or equivalently, Q < 13).
The user guide (Appendix B, page 122) for version 1.4 of the Illumina pipeline states that: "The scores are defined as
[11] Also, in Illumina runs using PhiX controls, the character 'B' was observed to represent an "unknown quality score".
For raw reads, the range of scores will depend on the technology and the base caller used, but will typically be up to 41 for recent Illumina chemistry.
[15] General-purpose tools such as Gzip and bzip2 regard FASTQ as a plain text file and result in suboptimal compression ratios.
General FASTQ compressors typically compress distinct fields (read names, sequences, comments, and quality scores) in a FASTQ file separately; these include DSRC and DSRC2, FQC, LFQC, Fqzcomp, and Slimfastq.
Reordering-based FASTQ compressors first cluster reads that share long substrings and then independently compress reads in each cluster after reordering them or assembling them into longer contigs, achieving perhaps the best trade-off between the running time and compression rate.
[18] Quality values account for about half of the required disk space in the FASTQ format (before compression), and therefore the compression of the quality values can significantly reduce storage requirements and speed up analysis and transmission of sequencing data.
For example, the algorithm QualComp[19] performs lossy compression with a rate (number of bits per quality value) specified by the user.
Based on rate-distortion theory results, it allocates the number of bits so as to minimize the MSE (mean squared error) between the original (uncompressed) and the reconstructed (after compression) quality values.
[21] Both are lossless compression algorithms that provide an optional controlled lossy transformation approach.
For example, SCALCE reduces the alphabet size based on the observation that “neighboring” quality values are similar in general.