Sequence alignment
These also include efficient, heuristic algorithms or probabilistic methods designed for large-scale database search, that do not guarantee to find best matches.
The Smith–Waterman algorithm is a general local alignment method based on the same dynamic programming scheme but with additional choices to start and end at any place.
Essential needs for an efficient and accurate method for DNA variant discovery demand innovative approaches for parallel processing in real time.
Optical computing approaches have been suggested as promising alternatives to the current electrical implementations, yet their applicability remains to be tested [1].
One way of quantifying the utility of a given pairwise alignment is the 'maximal unique match' (MUM), or the longest subsequence that occurs in both query sequences.
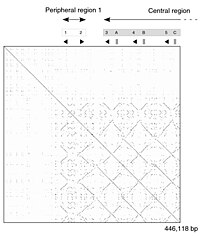
More precisely: "Given two genomes A and B, Maximal Unique Match (MUM) substring is a common substring of A and B of length longer than a specified minimum length d (by default d= 20) such that The dot-matrix approach, which implicitly produces a family of alignments for individual sequence regions, is qualitative and conceptually simple, though time-consuming to analyze on a large scale.
In the absence of noise, it can be easy to visually identify certain sequence features—such as insertions, deletions, repeats, or inverted repeats—from a dot-matrix plot.
Some implementations vary the size or intensity of the dot depending on the degree of similarity of the two characters, to accommodate conservative substitutions.
(In standard dynamic programming, the score of each amino acid position is independent of the identity of its neighbors, and therefore base stacking effects are not taken into account.
[10][11] Dynamic programming can be useful in aligning nucleotide to protein sequences, a task complicated by the need to take into account frameshift mutations (usually insertions or deletions).
In practice, the method requires large amounts of computing power or a system whose architecture is specialized for dynamic programming.
Only if this region is detected do these methods apply more sensitive alignment criteria; thus, many unnecessary comparisons with sequences of no appreciable similarity are eliminated.
The method is slower but more sensitive at lower values of k, which are also preferred for searches involving a very short query sequence.
Such conserved sequence motifs can be used in conjunction with structural and mechanistic information to locate the catalytic active sites of enzymes.
Although this technique is computationally expensive, its guarantee of a global optimum solution is useful in cases where only a few sequences need to be aligned accurately.
One method for reducing the computational demands of dynamic programming, which relies on the "sum of pairs" objective function, has been implemented in the MSA software package.
Many variations of the Clustal progressive implementation[15][16][17] are used for multiple sequence alignment, phylogenetic tree construction, and as input for protein structure prediction.
A variety of general optimization algorithms commonly used in computer science have also been applied to the multiple sequence alignment problem.
Hidden Markov models have been used to produce probability scores for a family of possible multiple sequence alignments for a given query set; although early HMM-based methods produced underwhelming performance, later applications have found them especially effective in detecting remotely related sequences because they are less susceptible to noise created by conservative or semiconservative substitutions.
The Burrows–Wheeler transform has been successfully applied to fast short read alignment in popular tools such as Bowtie and BWA.
[23] It can generate pairwise or multiple alignments and identify a query sequence's structural neighbors in the Protein Data Bank (PDB).
It has been extended since its original description to include multiple as well as pairwise alignments,[24] and has been used in the construction of the CATH (Class, Architecture, Topology, Homology) hierarchical database classification of protein folds.
A web-based server implementing the method and providing a database of pairwise alignments of structures in the Protein Data Bank is located at the Combinatorial Extension website.
Therefore, it does not account for possible difference among organisms or species in the rates of DNA repair or the possible functional conservation of specific regions in a sequence.
More statistically accurate methods allow the evolutionary rate on each branch of the phylogenetic tree to vary, thus producing better estimates of coalescence times for genes.
[36] The choice of a scoring function that reflects biological or statistical observations about known sequences is important to producing good alignments.
It can be very useful and instructive to try the same alignment several times with different choices for scoring matrix and/or gap penalty values and compare the results.
[40] The methods used for biological sequence alignment have also found applications in other fields, most notably in natural language processing and in social sciences, where the Needleman-Wunsch algorithm is usually referred to as Optimal matching.
[43] Business and marketing research has also applied multiple sequence alignment techniques in analyzing series of purchases over time.
[50][51] A comprehensive list of BAliBASE scores for many (currently 12) different alignment tools can be computed within the protein workbench STRAP.

Sequences are the amino acids for residues 120-180 of the proteins. Residues that are conserved across all sequences are highlighted in grey. Below the protein sequences is a key denoting conserved sequence (*), conservative mutations (:), semi-conservative mutations (.), and non-conservative mutations ( ). [ 2 ]