Transfer hydrogenation
Transfer hydrogenation usually occurs at mild temperature and pressure conditions using organic or organometallic catalysts, many of which are chiral, allowing efficient asymmetric synthesis.
It uses hydrogen donor compounds such as formic acid, isopropanol or dihydroanthracene, dehydrogenating them to CO2, acetone, or anthracene respectively.
A large scale application of transfer hydrogenation is coal liquefaction using "donor solvents" such as tetralin.
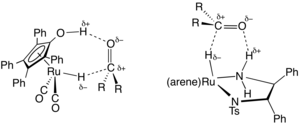
[2][3] In the area of organic synthesis, a useful family of hydrogen-transfer catalysts have been developed based on ruthenium and rhodium complexes, often with diamine and phosphine ligands.
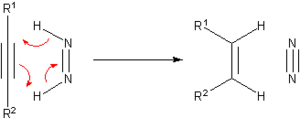
Transfer hydrogenations can proceed with high enantioselectivities when the starting material is prochiral: where RR'C*H−OH is a chiral product.
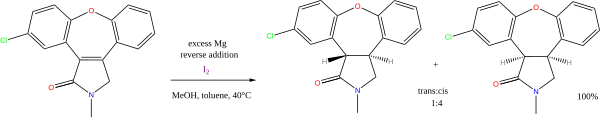
The combination of magnesium and methanol is used in alkene reductions, e.g. the synthesis of asenapine:[11] Organocatalytic transfer hydrogenation has been described by the group of List in 2004 in a system with a Hantzsch ester as hydride donor and an amine catalyst:[12]
By adopting a chiral imidazolidinone MacMillan organocatalyst an enantioselectivity of 81% ee was obtained: [13] In a case of stereoconvergence, both the E-isomer and the Z-isomer in this reaction yield the (S)-enantiomer.