Nitrone-olefin (3+2) cycloaddition
The cycloadditions is stereospecific with respect to the configuration of the alkene; however, diastereoselectivity in reactions of C-substituted nitrones is often low.
The (3+2) cycloaddition itself is a concerted, pericyclic process whose regiochemistry is controlled by the frontier molecular orbitals on the nitrone (the dipole) and the dipolarophile.
[3] When R' is an electron-donating group, alkyl, or aryl, the dominant FMOs are the HOMO of the dipolarophile and the LUMO of the nitrone.
Consistent with FMO control of the reaction, the more electron-withdrawing substituent on these substrates ends up in the 4 position of the product.
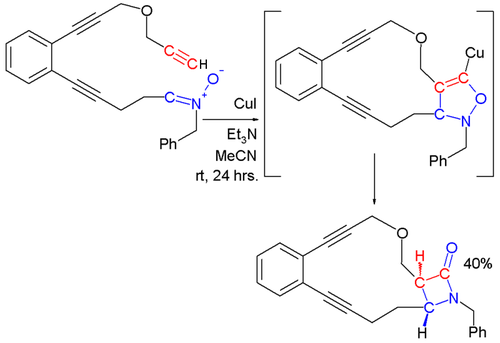
Regiochemistry is more difficult to predict for intramolecular reactions: either bridged or fused products can result, and both cis- and trans-fused rings are possible.
[9] The structure of hydroxycotinine, a human metabolite of nicotine, was confirmed via an independent synthesis employing nitrone-olefin cycloaddition.