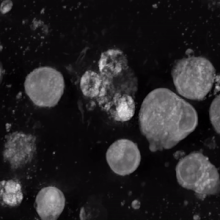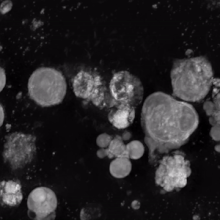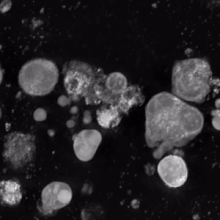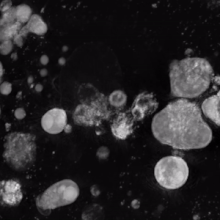
Megakaryocyte
In general, megakaryocytes are 10 to 15 times larger than a typical red blood cell, averaging 50–100 μm in diameter.
During its maturation, the megakaryocyte grows in size and replicates its DNA without cytokinesis in a process called endomitosis.
As a result, the nucleus of the megakaryocyte can become very large and lobulated, which, under a light microscope, can give the false impression that there are several nuclei.
TPO is sufficient but not absolutely necessary[2] for inducing differentiation of progenitor cells in the bone marrow towards a final megakaryocyte phenotype.
Many of the morphological features of megakaryocyte differentiation can be recapitulated in non-hematopoietic cells by the expression of Class VI β-tubulin (β6) and they provide a mechanistic basis for understanding these changes.
The maturation process occurs via endomitotic synchronous replication whereby the cytoplasmic volume enlarges as the number of chromosomes multiplies without cellular division.
TPO is primarily synthesized in the liver[8] but can be made by kidneys, testes, brain, and even bone marrow stromal cells.
The 2016 WHO requirements for diagnosis include > 450,000 platelets/μL of blood (normal 150,000–400,000) and characteristic findings in a bone marrow biopsy.
[12] Approximately half ET cases are due to a mutation in the JAK2 protein, a member of the JAK-STAT signaling pathway.
[15][16] In addition, there may be abnormalities with the central nervous system including the cerebrum and cerebellum that could cause symptoms.
There appears to be no generic resource for CAMT patients on the web and this is potentially due to the rarity of the disease.
In 1906, James Homer Wright provided evidence that megakaryocytes give rise to blood platelets.