Henry reaction
Discovered in 1895 by the Belgian chemist Louis Henry (1834–1913), it is the combination of a nitroalkane and an aldehyde or ketone in the presence of a base to form β-nitro alcohols.
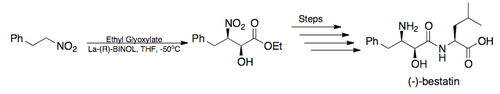
Many of these uses have been exemplified in the syntheses of various pharmaceuticals including the β-blocker (S)-propranolol,[5][6] the HIV protease inhibitor Amprenavir (Vertex 478), and construction of the carbohydrate subunit of the anthracycline class of antibiotics, L-Acosamine.
The Henry reaction begins with the deprotonation of the nitroalkane on the α-carbon position forming a nitronate.
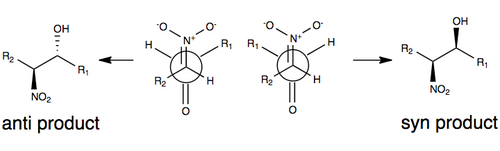
The figure below illustrates one of the commonly accepted models for stereoselection without any modification to the Henry reaction.
[3] In recent years, research focus has shifted toward modifications of the Henry reaction to overcome this synthetic challenge.
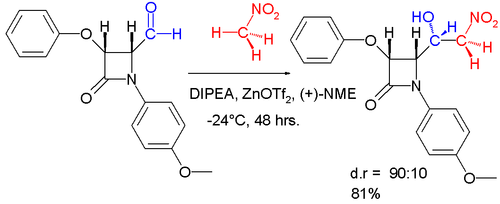
Of these some of the most important include employing high-pressure and sometimes solvent free conditions to improve chemo- and regioselectivity[2] and chiral metal catalysts to induce enantio-or diastereoselectivity.
[12] The aza-Henry reaction is also used to produce nitroamines and can be a reliable synthetic route for the synthesis of vicinal diamines.
This includes the conversion of unreactive alkyl nitro compounds to their corresponding dianions which will react faster with carbonyl substrates, reactions can be accelerated using PAP as base, utilization of the reactivity of aldehydes with α,α-doubly deprotonated nitroalkanes to give nitronate alkoxides that yield mainly syn-nitro alcohols once protonated, and finally generation of nitronate anions in which one oxygenatom on the nitro group is silyl-protected to yield anti-β-nitro alcohols in the presence of a fluoride anion source when reacted with an aldehyde.