Rubber elasticity
Natural rubbers, such as polybutadiene and polyisoprene, are extracted from plants as a fluid colloid and then solidified in a process called Vulcanization.
[5] By the mid-nineteenth century, the theory of thermodynamics was being developed and within this framework, the English mathematician and physicist Lord Kelvin[6] showed that the change in mechanical energy required to stretch a rubber sample should be proportional to the increase in temperature.
The connection to thermodynamics was firmly established in 1859 when the English physicist James Joule published the first careful measurements of the temperature increase that occurred as a rubber sample was stretched.
In 1838 the American inventor Charles Goodyear found that natural rubber's elastic properties could be immensely improved by adding a small amount of sulfur to produce chemical cross-links between adjacent polyisoprene molecules.
Before it is cross-linked, the liquid natural rubber consists of very long polymer molecules, containing thousands of isoprene backbone units, connected head-to-tail (commonly referred to as chains).
Because of the enormous economic and technological importance of rubber, predicting how a molecular network responds to mechanical strains has been of enduring interest to scientists and engineers.
To understand the elastic properties of rubber, theoretically, it is necessary to know both the physical mechanisms that occur at the molecular level and how the random-walk nature of the polymer chain defines the network.
The concept of entropy comes to us from the area of mathematical physics called statistical mechanics which is concerned with the study of large thermal systems, e.g. rubber networks at room temperature.
One may regard the entropic forces in polymer chains as arising from the thermal collisions that their constituent atoms experience with the surrounding material.
When these elastic force models are combined with the complex morphology of the network, it is not possible to obtain simple analytic formulae to predict the macroscopic stress.
It is only via numerical simulations on computers that it is possible to capture the complex interaction between the molecular forces and the network morphology to predict the stress and ultimate failure of a rubber sample as it is strained.
Over time scales of seconds to minutes, only these relatively short sections of the chain (i.e. kinks) have sufficient volume to move freely amongst their possible rotational conformations.
[10] The third mechanism occurs at high chain extension, as it is extended beyond its initial equilibrium contour length by the distortion of the chemical bonds along its backbone.
[11] The three force mechanisms are found to roughly correspond to the three regions observed in tensile stress vs. strain experiments, shown in Fig.
As the initial strain is applied to the rubber sample, the network nodes at the ends of the chain begin to move apart and all of the kink vectors along the contour are stretched simultaneously.
Physically, the applied strain forces the kinks to stretch beyond their thermal equilibrium end-to-end distances, causing a decrease in their entropy.
The force constant for the low strain regime can be estimated by sampling molecular dynamics (MD) trajectories of a kink (i.e. short chains) composed of 2–3 isoprene units, at relevant temperatures (e.g.
Since these distributions (which turn out to be approximately Gaussian) are directly related to the number of states, they may be associated with the entropy of the kink at any end-to-end distance.
By numerically differentiating the probability distribution, the change in entropy, and hence free energy, with respect to the kink end-to-end distance can be found.
[14][15][16] Using the joint probability distribution in equation (4) and the force extension models, it is possible to devise numerical algorithms to both construct a faithful representative volume element of a network and to simulate the resulting mechanical stress as it is subjected to strain.
When the force constant obtained for kinks having 2 or 3 isoprene units (approximately one Kuhn length) is used in numerical simulations, the predicted stress is found to be consistent with experiments.
These simulations also predict a steep upturn in the stress as network chains become taut and, ultimately, material failure due to bond rupture.
At low to moderate strains, theory predicts that the required stretching force is due to a change in entropy in the network chains.
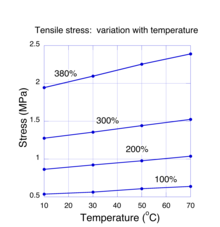
The positive linear behaviour of the stress with temperature sometimes leads to the mistaken notion that rubber has a negative coefficient of thermal expansion (i.e. the length of a sample shrinks when heated).
When one end of the sample is released, it snaps back to its original length too quickly for the naked eye to resolve the process.
Then, as the free end of the rubber snapped back, the styli traced out helical paths in the lamp black coating of the rotating cylinder.
The linear behaviour of the displacement vs. time indicates that, after a brief acceleration, both the end and the midpoint of the sample snapped back at a constant velocity of about 50 m/s or 112 mph.
Since T is always positive (it can never reach absolute zero), the ΔS must be negative, implying that the rubber in its natural state is more entangled (with more microstates) than when it is under tension.
This last phenomenon is the critical clue that the ability of an elastomer to do work depends (as with an ideal gas) only on entropy-change considerations, and not on any stored (i.e. potential) energy within the polymer bonds.
By using the following basic equations for Helmholtz free energy and its discussion about entropy, the force generated from the deformation of a rubber chain from its original unstretched conformation can be derived.