Selective progesterone receptor modulator
This mixed profile of action leads to stimulation or inhibition in tissue-specific manner, which further raises the possibility of dissociating undesirable adverse effects from the development of synthetic PR-modulator drug candidates.
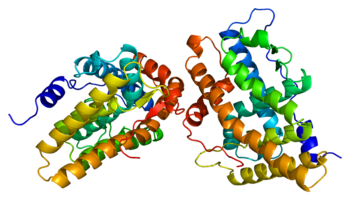
[20][21] The ligand binding site, known as AF2 domain, is expressed by exons 4-8, corresponding to 253 amino acids, and its structure is of great interest to SPRM development.
[25] At the turn of the millennium it was apparent that progesterone activity was not mediated only via transcription factor, but also by a membrane-bound G protein-coupled receptor designated as 7TMPR.
When the receptor is activated it blocks adenylyl cyclase, leading to decreased biosynthesis of the intracellular second-messenger cAMP.
[12] Studies have shown that PR-B activation is important for growth and development of the mammary gland, whereas PR-A has a significant role in normal reproductive function and ovulation.
[26] Studies comparing the conformational changes in helix-12 contributing to agonistic and antagonistic effects have shown an important hydrogen interaction with Glu723 residue of helix-3.
However, when an antagonist, e.g. mifepristone, interacts with this hydrogen bond system then its dimethylamine group clashes in to Met909 and destabilizes helix-12, causing a conformational change, which promotes the recruitment of corepressors.
[26][28] When SPRMs bind to the progesterone receptor, the equilibrium between the two conformational states is more closely balanced and hence more easily perturbed by differences in the cellular environment.
In the nucleus the dimer interacts with progesterone hormone response element in the DNA causing upregulation or downregulation of the gene.
[26] In the action of agonism there occur conformational changes, where alpha helices 3, 4 and 12 create a docking surface for coactivator proteins, which act as bridging factors between the receptor and the general transcription machinery.
[37][38] However, the antagonist prevents proper packing of alpha helix 12 against helices 3 and 4, impairing the receptor’s ability to interact with coactivators, which allows recruitment of corepressor, such as SMRT and NCoR.
[40] The selective progesterone receptor modulators have been described as agents with mixed agonist-antagonist activity and thus the mechanism of action must be due to a balance of these functions.
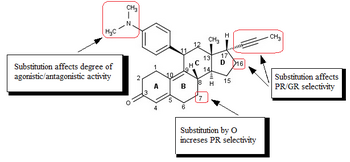
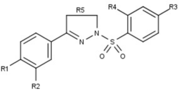
[41][42][43] Minor changes in the 17-alpha region generate antiprogestins with reduced antiglucocorticoidal activity, where alpha refers to an absolute steroidal stereodescriptor.
[43] Substitution on the 17-alpha position involving phenyl group with small, electron-withdrawing substituents, such as F and CF3, on the para-position was also found to greatly increase the selectivity over glucocorticoid receptor as well as the potency of resulting compounds.
[41][42] Small substituents like methyl or vinyl give rise to potent progesterone receptor-agonistic properties[42] whereas substituted phenyl derivatives show different degrees of antagonistic activity.
4) by oxygen atom has been investigated and these mifepristone-like oxasteroids showed increased selectivity over glucocorticoid receptor but were less potent than mifepristone.
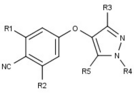
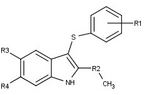
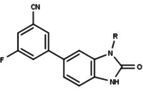
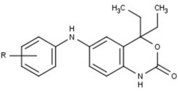
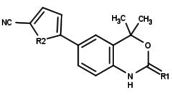
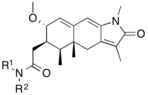
Variety of new types of progesterone receptor antagonists with different degree of potency has been reported and show a remarkable structural diversity which can be seen in table below.
In contrast to conventional progesterone antagonists, the SPRMs eliminate the ability to terminate pregnancy due to their mixed antagonist/agonist profile.
[50] In post-menopausal endometrium the compound seems to have antagonistic effect or progesterone receptor, indicating potential use in menopausal treatment but this has yet to be confirmed.
[60] Due to its antiglucocorticoidal activity, mifepristone is investigated for its therapeutical potential in indications like Cushing's syndrome, Alzheimer's disease or psychosis.