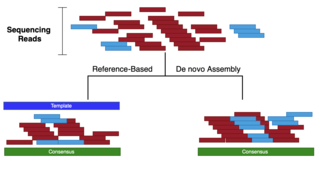
Sequence assembly
[1] Typically, the short fragments (reads) result from shotgun sequencing genomic DNA, or gene transcript (ESTs).
Reads in each group will then be reduced in size using the k-mere approach to select the highest quality and most probable contiguous (contig).
[2] As the sequenced organisms grew in size and complexity (from small viruses over plasmids to bacteria and finally eukaryotes), the assembly programs used in these genome projects needed increasingly sophisticated strategies to handle: Faced with the challenge of assembling the first larger eukaryotic genomes—the fruit fly Drosophila melanogaster in 2000 and the human genome just a year later,—scientists developed assemblers like Celera Assembler[4] and Arachne[5] able to handle genomes of 130 million (e.g., the fruit fly D. melanogaster) to 3 billion (e.g., the human genome) base pairs.
Subsequent to these efforts, several other groups, mostly at the major genome sequencing centers, built large-scale assemblers, and an open source effort known as AMOS[6] was launched to bring together all the innovations in genome assembly technology under the open source framework.
The input sequences for EST assembly are fragments of the transcribed mRNA of a cell and represent only a subset of the whole genome.
EST assembly is made much more complicated by features like (cis-) alternative splicing, trans-splicing, single-nucleotide polymorphism, and post-transcriptional modification.
Beginning in 2008 when RNA-Seq was invented, EST sequencing was replaced by this far more efficient technology, described under de novo transcriptome assembly.
[10] From 2006, the Illumina (previously Solexa) technology has been available and can generate about 100 million reads per run on a single sequencing machine.
Compare this to the 35 million reads of the human genome project which needed several years to be produced on hundreds of sequencing machines.
[11] Illumina was initially limited to a length of only 36 bases, making it less suitable for de novo assembly (such as de novo transcriptome assembly), but newer iterations of the technology achieve read lengths above 100 bases from both ends of a 3-400bp clone.
Despite the higher error rates of these technologies they are important for assembly because their longer read length helps to address the repeat problem.
[14] The initial BUSCO sets represented 3023 genes for vertebrates, 2675 for arthropods, 843 for metazoans, 1438 for fungi and 429 for eukaryotes.