Hereditary spastic paraplegia
Hereditary spastic paraplegia (HSP) is a group of inherited diseases whose main feature is a progressive gait disorder.
The main feature of the disease is progressive spasticity in the lower limbs due to pyramidal tract dysfunction.
Furthermore, among the core symptoms of HSP are also included abnormal gait and difficulty in walking, decreased vibratory sense at the ankles, and paresthesia.
[8] Individuals with HSP can experience extreme fatigue associated with central nervous system and neuromuscular disorders, which can be disabling.
[13] More specifically, patients with the autosomal dominant pure form of HSP reveal normal facial and extraocular movement.
Although jaw jerk may be brisk in older subjects, there is no speech disturbance or difficulty of swallowing.
Weakness is most notable at the iliopsoas, tibialis anterior, and to a lesser extent, hamstring muscles.
Pure HSP presents with spasticity in the lower limbs, associated with neurogenic bladder disturbance as well as lack of vibration sensitivity (pallhypesthesia).
On the other hand, HSP is classified as complex when lower limb spasticity is combined with any additional neurological symptom.
[15] New findings propose that an earlier onset leads to a longer disease duration without loss of ambulation or the need for the use of a wheelchair.
[13] However, this is not always the case as De Novo Early Onset SPG4, a form of infantile HSP, involves loss of ambulation and other motor skills.
The gene locations are in the format: chromosome - arm (short or p: long or q) - band number.
Neuronal cell bodies of degenerating axons are preserved and there is no evidence of primary demyelination.
[3] Axons in the central and peripheral nervous system are coated with an insulation, the myelin layer, to increase the speed of action potential propagation.
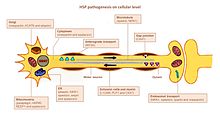
[citation needed] Endoplasmic reticulum (ER) is the main organelle for lipid synthesis.
Mutations in genes encoding proteins that have a role in shaping ER morphology and lipid metabolism were linked to HSP.
Dysfunction of endosomal trafficking can have severe consequences in motor neurons with long axons, as reported in HSP.
[citation needed] Initial diagnosis of HSPs relies upon family history, the presence or absence of additional signs and the exclusion of other nongenetic causes of spasticity, the latter being particular important in sporadic cases.
[7] Cerebral and spinal MRI is an important procedure performed in order to rule out other frequent neurological conditions, such as multiple sclerosis, but also to detect associated abnormalities such as cerebellar or corpus callosum atrophy as well as white matter abnormalities.
[citation needed] Hereditary spastic paraplegias can be classified based on the symptoms; mode of inheritance; the patient's age at onset; the affected genes; and biochemical pathways involved.
Available therapies mainly consist of symptomatic medical management and promoting physical and emotional well-being.
No differences in rate relating to gender were found, and average age at onset was 24 years.
[33] In the United States, Hereditary Spastic Paraplegia is listed as a "rare disease" by the Office of Rare Diseases (ORD) of the National Institutes of Health which means that the disorder affects less than 200,000 people in the US population.