Super-resolution microscopy
There are two major groups of methods for super-resolution microscopy in the far-field that can improve the resolution by a much larger factor:[10] On 8 October 2014, the Nobel Prize in Chemistry was awarded to Eric Betzig, W.E.
[11][12] The different modalities of super-resolution microscopy are increasingly being adopted by the biomedical research community, and these techniques are becoming indispensable tools to understanding biological function at the molecular level.
[14] However the publication from 1978 [15] had drawn an improper physical conclusion (i.e. a point-like spot of light) and had completely missed the axial resolution increase as the actual benefit of adding the other side of the solid angle.
In comparison with NSOM and ANSOM this method does not require any special equipment for tip positioning and has a large field of view and a depth of focus.
[26] Structured illumination microscopy (SIM) enhances spatial resolution by collecting information from frequency space outside the observable region.
Licenses to this technology were procured by Dyer Energy Systems, Calimetrics Inc., and Nanoptek Corp. for use of this super-resolution technique in optical data storage and microscopy.
Like standard structured illumination, the SMI technique modifies the point spread function (PSF) of a microscope in a suitable manner.
[32][33][34] SMI can be combined with other super resolution technologies, for instance with 3D LIMON or LSI-TIRF as a total internal reflection interferometer with laterally structured illumination (this last instrument and technique is essentially a phase-shifted photon tunneling microscope, which employs a total internal reflection light microscope with phase-shifted evanescent field (Guerra, 1996).
[31] This SMI technique allows one to acquire light-optical images of autofluorophore distributions in sections from human eye tissue with previously unmatched optical resolution.
Various microscopy methods, including super-resolution optical fluctuation imaging, have been used to quantify and monitor biological activities in real time.
[48] Saturated structured-illumination microscopy (SSIM) exploits the nonlinear dependence of the emission rate of fluorophores on the intensity of the excitation laser.
[49] By applying a sinusoidal illumination pattern[50] with a peak intensity close to that needed in order to saturate the fluorophores in their fluorescent state, one retrieves Moiré fringes.
In addition there is the need for very photostable fluorophores, due to the saturating conditions, which inflict radiation damage on the sample and restrict the possible applications for which SSIM may be used.
Single-molecule localization microscopy (SMLM) summarizes all microscopical techniques that achieve super-resolution by isolating emitters and fitting their images with the point spread function (PSF).
Suitable fluorophores (e.g. for STORM) reside in a non-fluorescent dark state for most of the time and are activated stochastically, typically with an excitation laser of low intensity.
The photons emitted during the fluorescent phase are collected with a camera and the resulting image of the fluorophore (which is distorted by the PSF) can be fitted with very high precision, even on the order of a few Angstroms.
[54] The localization precision in this approach is enhanced because the slower photochemistry at low temperatures leads to a higher number of photons that can be emitted from each fluorophore before photobleaching.
[63][64] Consequently, cryogenic stochastic localization microscopy achieves the sub-molecular resolution required to resolve the 3D positions of several fluorophores attached to a small protein.
The method can also be combined with other structural biology techniques—such as X-ray crystallography, magnetic resonance spectroscopy, and electron microscopy—to provide valuable complementary information and specificity.
[65] By careful adjustment of the chemical environment—leading to local, reversible DNA melting and hybridization control over the fluorescence signal—DNA-binding dye molecules can be introduced.
[70][71][85][86] Highly specific labeling of biological structures with photoswitchable probes has been achieved with antibody staining,[81][82][83][87] direct conjugation of proteins,[88] and genetic encoding.
[70][71][85][86] STORM has also been extended to three-dimensional imaging using optical astigmatism, in which the elliptical shape of the point spread function encodes the x, y, and z positions for samples up to several micrometers thick,[82][87] and has been demonstrated in living cells.
[89][90] The first dye used was Nile red which is nonfluorescent in aqueous solution but fluorescent when inserted into a hydrophobic environment, such as micelles or living cell walls.
The stochastic binding of single-dye molecules (probes) to an immobilized target can be spatially and temporally resolved under a typical widefield fluorescence microscope.
However, when it binds to a fixed target, the dye stops moving; and clear input into the point spread function can be achieved.
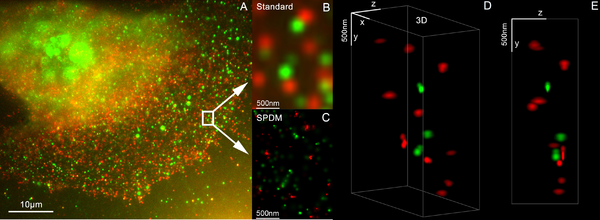
By using two different laser wavelengths, SPDM reveals cellular objects which are not detectable under conventional fluorescence wide-field imaging conditions, beside making for a substantial resolution improvement of autofluorescent structures.
[100] Label-free superresolution microscopy has also been demonstrated using the fluctuations of a surface-enhanced Raman scattering signal on a highly uniform plasmonic metasurface.
[105] Localization microscopy depends heavily on software that can precisely fit the point spread function (PSF) to millions of images of active fluorophores within a few minutes.
[108] It is possible to circumvent the need for PSF fitting inherent in single molecule localization microscopy (SMLM) by directly computing the temporal autocorrelation of pixels.
This technique is called super-resolution optical fluctuation imaging (SOFI) and has been shown to be more precise than SMLM when the density of concurrently active fluorophores is very high.