Thioredoxin reductase
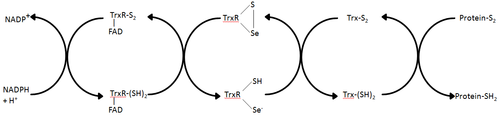
Each monomer contains a FAD prosthetic group, a NADPH binding domain, and an active site containing a redox-active disulfide bond.
Electrons are taken from NADPH via TrxR and are transferred to the active site of Trx, which goes on to reduce protein disulfides or other substrates.
Mammalian ThxR has an insertion in the FAD binding domain between two alpha helices, which forms a small pair of beta strands.
An additional feature of the mammalian mechanism is the presence of a selenocysteine residue at the C-terminal end of the protein which is required for catalytic activity.
Since the activity of this enzyme is essential for cell growth and survival, it is a good target for anti-tumor therapy.
Thioredoxin reductases are essential proteins for regulating cellular redox balance and mitigating the damage caused by reactive oxygen species generated via oxidative phosphorylation in the mitochondria.
Inactivation of mitochondrial TrxR2 in mice results in thinning of the ventricular heart walls and neonatal death.
It is hypothesized that the pathological impact of these mutations is an impaired ability to control oxidative damage in cardiac myocytes.
[24] There has recently been some research to show that low molecular weight thioredoxin reductase could be a target for novel antibiotics (such as auranofin or Ebselen.