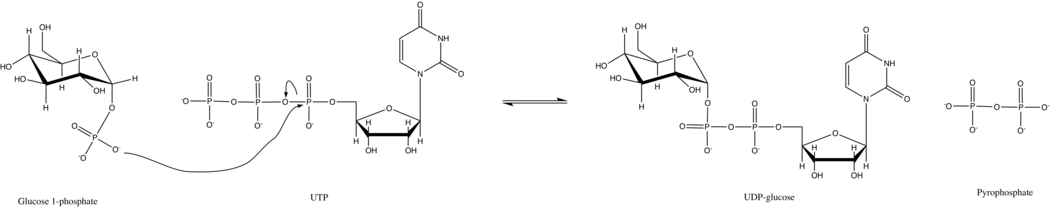
UTP—glucose-1-phosphate uridylyltransferase
It synthesizes UDP-glucose from glucose-1-phosphate and UTP; i.e., UTP—glucose-1-phosphate uridylyltransferase is an enzyme found in all three domains (bacteria, eukarya, and archaea) as it is a key player in glycogenesis and cell wall synthesis.
[5][6] In humans and in yeast, the enzyme is active as an octamer consisting of two tetramers stacked onto one another with conserved hydrophobic residues at the interfaces between the subunits.
A Rossman fold motif participates in binding of the UTP nucleotide and a sugar-binding domain (residues T286–G293) coordinates with the glucose ring.
[11] UTP—glucose-1-phosphate uridylyltransferase is ubiquitous in nature due to its important role in the generation of UDP-glucose, a central compound in carbohydrate metabolism.
In plant leaves, UTP—glucose-1-phosphate uridylyltransferase is a key part of the sucrose biosynthesis pathway, supplying Uridine diphosphate glucose to Sucrose-phosphate synthase which converts UDP-glucose and D-fructose 6-phosphate into sucrose-6-phosphate.
In higher animals, the enzyme is highly active in tissues involved in glycogenesis, including the liver and the muscles.
However, under typical cellular conditions, inorganic pyrophosphatase quickly hydrolyzes the pyrophosphate product and drives the reaction forward by Le Chatelier's Principle.
[8] Similar to other sugar nucleotidyltransferases, UTP—glucose-1-phosphate uridylyltransferase activity requires two divalent cations to stabilize the binding of negatively charged phosphate groups.
[5] In addition to stabilizing the negatively charged phosphates, Mg2+ is thought to orient the glucose 1-phosphate for nucleophilic attack of the α-phosphorus of UTP.
Indeed, UGP2 expression is increased in response to stressors such as phosphate deficiency, indicating that UGP2 probably functions as a backup to UGP1 when the plant is under environmental stress.
[3] UDP-glucose pyrophosphorylase (UGP2) was recently found to be implicated in novel neurodevelopmental disorder in humans, known as [32] also referred to as Barakat-Perenthaler syndrome.
[33] This disorder was first described in 22 individuals from 15 families, presenting with a severe epileptic encephalopathy, neurodevelopmental delay with absence of virtually all developmental milestones, intractable seizures, progressive microcephaly, visual disturbance and similar minor dysmorphisms.
Functional studies from the same group showed that the short protein isoform is normally predominantly expressed in human brain.