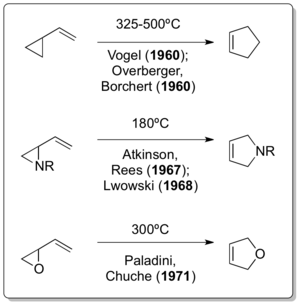
Vinylcyclopropane rearrangement
Experimental and computational investigations show that mechanistically, the vinylcyclopropane rearrangement can be thought of as either a diradical-mediated two-step and/or orbital-symmetry-controlled pericyclic process.
Due to its ability to form cyclopentene rings the vinylcyclopropane rearrangement has served several times as a key reaction in complex natural product synthesis.
In 1959, a young research chemist with Humble Oil and Refining (Esso, now Exxon) named Norman P. Neureiter was instructed to find new uses for the excess butadiene produced from one of the refinery processes.
[8] The corresponding all-carbon version of the reaction was independently reported by Emanuel Vogel[9] and Overberger & Borchert just one year after the Neureiter publication appeared.
[21] The kinetic data obtained for this rearrangement were consistent with a concerted mechanism where cleavage of the cyclopropyl carbon-carbon bond was rate-limiting.
[23] Immediately people started to appreciate the possibility for a diradical intermediate arising from homolytic cleavage of the weak C1-C2-cyclopropane bond under thermal conditions.
Woodward and Hoffmann based their analysis solely on the principles of the conservation of orbital symmetry theory without however making any mechanistic or stereochemical prediction.
The attention directed towards the vinylcyclopropane rearrangement by Woodward and Hoffmann as a representative example for [1,3]-carbon shifts clearly enhanced the interest in this reaction.
Substituents with increased radical stabilizing ability not only lower the rearrangements activation energy but also reclosure of the initially formed diradical species becomes slower relative to the rate of cyclopentene formation resulting in an overall more concerted mechanism with less stereomutation (e.g. entry 6 & 7).
The data is consistent with the formation of biradical species on a relatively flat potential energy surface allowing for restricted conformational flexibility before the products are formed.
Not only do these high temperatures allow side reactions with similar activation energies, such as homodienyl-[1,5]-hydrogen shifts, to occur but also do they significantly limit the functional groups tolerated in the substrates.
Even though the dithiane-substituted vinylcyclopropane substrates required two synthetic steps starting from the corresponding 1,3-dienes the method proved itself successful for the synthesis of a variety of substituted cyclopentenes.
In contrast to the larger, fully "consonant" cyclohexane scaffold cyclopentanes and their derivatives are "dissonant" according to the Lapworth-Evans model of alternating polarities.
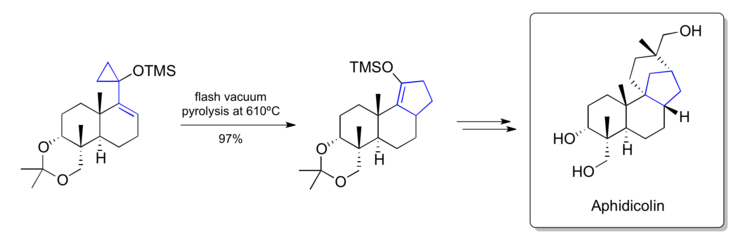
[40] A key step converts a late stage siloxyvinylcyclopropane into a cyclopentene that contained the [6-6-5]-fused carbon skeleton found within the natural product.
[41] The methodology has also been applied to the synthesis hirsutene[42] and isocomene[43] Cinylcyclopropane rearrangement has been used to build the spirocyclic natural product alpha-vetispirene in 1982.