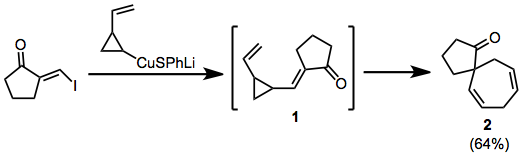
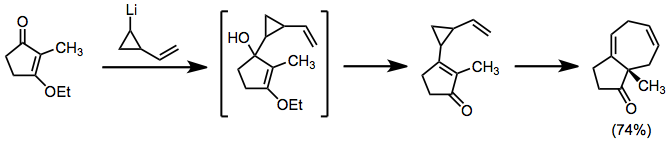
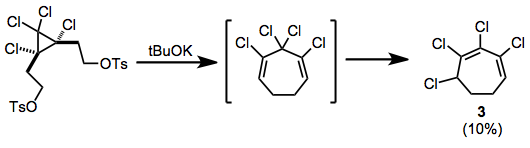
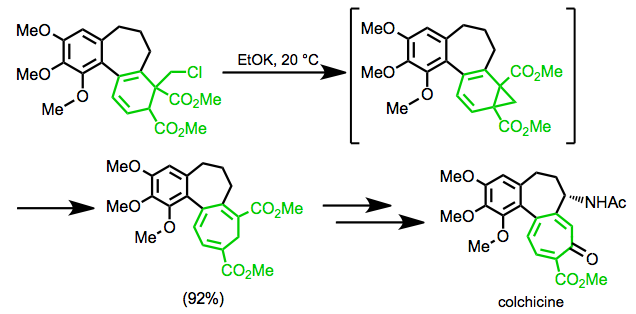
Divinylcyclopropane-cycloheptadiene rearrangement
It is conceptually related to the Cope rearrangement, but has the advantage of a strong thermodynamic driving force due to the release of ring strain.
Disadvantages: The configuration of the starting materials needs be controlled in many cases—trans-divinylcyclopropanes often require heating to facilitate isomerization before rearrangement will occur.
Mechanistic experiments have shown that trans-divinylcyclopropanes epimerize to the corresponding cis isomers and undergo the rearrangement via what is most likely a concerted pathway.
In one example employing rhodium bis(ethylene) hexafluoroacetylacetonate, coordination and formation of a bis-π-allyl complex precede electrocyclic ring closure and catalyst release.
[8] (6)Rearrangement after elimination of ditosylates has been observed; the chlorinated cycloheptadiene thus produced isomerizes to conjugated heptadiene 3 during the reaction.
[14] Conjugated dienyl epoxides form similar products, lending support to the existence of an ylide intermediate.
The earliest observation of a cycloheptadiene via the title rearrangement was made by Baeyer in his synthesis of eucarvone from carvone hydrobromide.
The solution was stirred at –78° for 15 minutes and at room temperature for 2–3 hours, and then it was partitioned between saturated aqueous sodium bicarbonate and pentane (10 mL and 20 mL/mmol of ketone, respectively).
Thermolysis of the silyl enol ether was accomplished by heating (neat, argon atmosphere) at 230° (air-bath temperature) for 30–60 minutes.
Direct distillation (140–150°/12 torr) of the resultant materials provided the cycloheptadiene in 85% yield: IR (film) 1660, 1260, 840 cm–1; 1H NMR (CDCl3) δ 0.09 (s, 6H), 0.88 (s, 9H), 0.7–2.75 (m, 14H), 4.8 (t, 1H, J = 5.5 Hz), 5.5–5.9 (m, 2H).