Adenine phosphoribosyltransferase
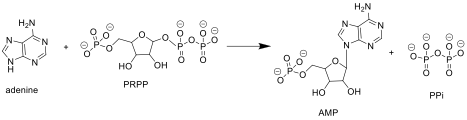
[8] APRTase catalyzes the following reaction in the purine nucleotide salvage pathway: Adenine + Phosphoribosyl Pyrophosphate (PRPP) → Adenylate (AMP) + Pyrophosphate (PPi) In organisms that can synthesize purines de novo, the nucleotide salvage pathway provides an alternative that is energetically more efficient.
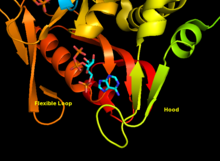
Although the flexible loop does not interact with the hood during purine recognition, it is thought to close over the active site and sequester the reaction from solvents.
[12] However, a recent effort to resolve the structure of human APRTase was unable to locate a single site for Mg2+, but did find evidence to suggest a Cl− atom near Trp98.
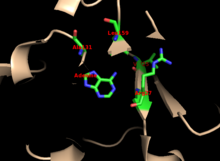
[9] In human APRTase, it is thought that adenine's N9 proton is abstracted by Glu104 to form an oxacarbenium transition state.
The mechanism of APRTase is generally consistent with that of other PRTases, which conserve the function of displacing PRPP's α-1-pyrophosphate using a nitrogen nucleophile, in either an SN1 or SN2 attack.
[14] Type I deficiency results in a complete loss of APRTase activity and can occur in patients that are homozygous or compound heterozygous for various mutations.
[17] These mutations cause effects that are clustered into three main areas: in the binding of PRPP's β-phosphate, in the binding of PRPP's 5'-phosphate, and in the segment of the flexible loop that closes over the active site during catalysis [10] Type I deficiency has been observed in various ethnic groups but studied predominately among White populations.
[17] Type II deficiency causes APRTase to have a reduced affinity for PRPP, resulting in a tenfold increase in the KM value.
It is treatable with regular doses of allopurinol or febuxostat, which inhibit xanthine dehydrogenase activity to prevent the accumulation and precipitation of DHA.