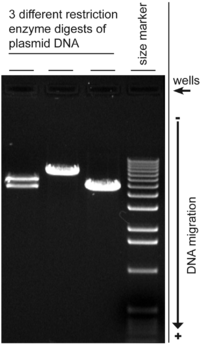
Agarose gel electrophoresis
Agarose gel electrophoresis is a method of gel electrophoresis used in biochemistry, molecular biology, genetics, and clinical chemistry to separate a mixed population of macromolecules such as DNA or proteins in a matrix of agarose, one of the two main components of agar.
[2] Agarose gel is easy to cast, has relatively fewer charged groups, and is particularly suitable for separating DNA of size range most often encountered in laboratories, which accounts for the popularity of its use.
[3] The 3-D structure is held together with hydrogen bonds and can therefore be disrupted by heating back to a liquid state.
[7] It can also be used to separate large proteins, and it is the preferred matrix for the gel electrophoresis of particles with effective radii larger than 5–10 nm.
The lower sulfate content of low EEO agarose, particularly low-melting point (LMP) agarose, is also beneficial in cases where the DNA extracted from gel is to be used for further manipulation as the presence of contaminating sulfates may affect some subsequent procedures, such as ligation and PCR.
However, for some applications such as the electrophoresis of serum proteins, a high EEO may be desirable, and agaropectin may be added in the gel used.
[10] A number of factors can affect the migration of nucleic acids: the dimension of the gel pores (gel concentration), size of DNA being electrophoresed, the voltage used, the ionic strength of the buffer, and the concentration of intercalating dye such as ethidium bromide if used during electrophoresis.
However, the migration of DNA molecules in solution, in the absence of a gel matrix, is independent of molecular weight during electrophoresis.
This model assumes that the DNA can crawl in a "snake-like" fashion (hence "reptation") through the pores as an elongated molecule.
[21] Real-time fluorescence microscopy of stained molecules, however, showed more subtle dynamics during electrophoresis, with the DNA showing considerable elasticity as it alternately stretching in the direction of the applied field and then contracting into a ball, or becoming hooked into a U-shape when it gets caught on the polymer fibres.
[22][23] The details of an agarose gel electrophoresis experiment may vary depending on methods, but most follow a general procedure.
The loading buffer also includes colored dyes such as xylene cyanol and bromophenol blue used to monitor the progress of the electrophoresis.
It is also possible, but less common, to perform the electrophoresis vertically, as well as horizontally with the gel raised on agarose legs using an appropriate apparatus.
Too high a voltage may also reduce resolution, as well as causing band streaking for large DNA molecules.
Too low a voltage may lead to broadening of band for small DNA fragments due to dispersion and diffusion.
Less commonly used dyes include Cresol Red and Orange G which migrate ahead of bromophenol blue.
DNA stained with crystal violet can be viewed under natural light without the use of a UV transilluminator which is an advantage, however it may not produce a strong band.
Using a higher wavelength of 365 nm (UV-A range) causes less damage to the DNA but also produces much weaker fluorescence with ethidium bromide.
In general, the ideal buffer should have good conductivity, produce less heat and have a long life.
[31] There are a number of buffers used for agarose electrophoresis; common ones for nucleic acids include tris/acetate/EDTA (TAE) and tris/borate/EDTA (TBE).
; in most cases the purported rationale is lower current (less heat) and or matched ion mobilities, which leads to longer buffer life.
LB is relatively new and is ineffective in resolving fragments larger than 5 kbp; However, with its low conductivity, a much higher voltage could be used (up to 35 V/cm), which means a shorter analysis time for routine electrophoresis.
[33] Agarose gels are easily cast and handled compared to other matrices and nucleic acids are not chemically altered during electrophoresis.
Electrophoresis is performed in buffer solutions to reduce pH changes due to the electric field, which is important because the charge of DNA and RNA depends on pH, but running for too long can exhaust the buffering capacity of the solution.