Bayesian inference in phylogeny
Bayesian inference of phylogeny combines the information in the prior and in the data likelihood to create the so-called posterior probability of trees, which is the probability that the tree is correct given the data, the prior and the likelihood model.
Bayesian inference was introduced into molecular phylogenetics in the 1990s by three independent groups: Bruce Rannala and Ziheng Yang in Berkeley,[1][2] Bob Mau in Madison,[3] and Shuying Li in University of Iowa,[4] the last two being PhD students at the time.
Published posthumously in 1763 it was the first expression of inverse probability and the basis of Bayesian inference.
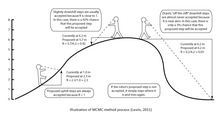
MCMC methods can be described in three steps: first using a stochastic mechanism a new state for the Markov chain is proposed.
The number of times a single tree is visited during the course of the chain is an approximation of its posterior probability.
The algorithm has two components: Metropolis-coupled MCMC algorithm (MC³) [12] has been proposed to solve a practical concern of the Markov chain moving across peaks when the target distribution has multiple local peaks, separated by low valleys, are known to exist in the tree space.
The (MC³) improves the mixing of Markov chains in presence of multiple local peaks in the posterior density.
After each iteration, a swap of states between two randomly chosen chains is proposed through a Metropolis-type step.
is ideally suited for implementation on parallel machines, since each chain will in general require the same amount of computation per iteration.
The two endpoints of the first branch selected will have a sub-tree hanging like a piece of clothing strung to the line.
The algorithm proceeds by multiplying the three selected branches by a common random amount, akin to stretching or shrinking the clothesline.
Finally the leftmost of the two hanging sub-trees is disconnected and reattached to the clothesline at a location selected uniformly at random.
There are many approaches to reconstructing phylogenetic trees, each with advantages and disadvantages, and there is no straightforward answer to “what is the best method?”.
Maximum Parsimony recovers one or more optimal trees based on a matrix of discrete characters for a certain group of taxa and it does not require a model of evolutionary change.
MP gives the most simple explanation for a given set of data, reconstructing a phylogenetic tree that includes as few changes across the sequences as possible.
For the same reason that it has been widely used, its simplicity, MP has also received criticism and has been pushed into the background by ML and Bayesian methods.
As shown by Felsenstein (1978), MP might be statistically inconsistent,[15] meaning that as more and more data (e.g. sequence length) is accumulated, results can converge on an incorrect tree and lead to long branch attraction, a phylogenetic phenomenon where taxa with long branches (numerous character state changes) tend to appear more closely related in the phylogeny than they really are.
For morphological data, recent simulation studies suggest that parsimony may be less accurate than trees built using Bayesian approaches,[16] potentially due to overprecision,[17] although this has been disputed.
However it considers the probability of each tree explaining the given data based on a model of evolution.
This approach might eliminate long branch attraction and explain the greater consistency of ML over MP.
Although considered by many to be the best approach to inferring phylogenies from a theoretical point of view, ML is computationally intensive and it is almost impossible to explore all trees as there are too many.
Bayesian inference also incorporates a model of evolution and the main advantages over MP and ML are that it is computationally more efficient than traditional methods, it quantifies and addresses the source of uncertainty and is able to incorporate complex models of evolution.
[28] As Bayesian methods increased in popularity, MrBayes became one of the software of choice for many molecular phylogeneticists.
MrBayes reads aligned matrices of sequences (DNA or amino acids) in the standard NEXUS format.
[9] The user can change assumptions of the substitution model, priors and the details of the MC³ analysis.
It offers different methods for relaxing the assumption of equal substitutions rates across nucleotide sites.
[31] MrBayes is also able to infer ancestral states accommodating uncertainty to the phylogenetic tree and model parameters.
This new framework allows the user to mix models and take advantages of the efficiency of Bayesian MCMC analysis when dealing with different type of data (e.g. protein, nucleotide, and morphological).
Version 3.2 provides wider outputs options compatible with FigTree and other tree viewers.
This table includes some of the most common phylogenetic software used for inferring phylogenies under a Bayesian framework.