Biological small-angle scattering
Small-angle scattering is used to study the structure of a variety of objects such as solutions of biological macromolecules, nanocomposites, alloys, and synthetic polymers.
In contrast to other X-ray and neutron scattering methods, SAS yields information on the sizes and shapes of both crystalline and non-crystalline particles.
Solid-State NMR is still an indispensable tool for determining atomic level information of macromolecules greater than 40 kDa or non-crystalline samples such as amyloid fibrils.
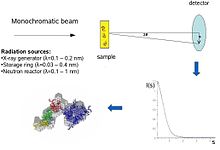
The biological SAXS method profits from the high intensity of X-ray photon beams provided by the synchrotron storage rings.
[1] The information content of SAS data is illustrated here in the figure on the right, which shows X-ray scattering patterns from proteins with different folds and molecular masses.
At low angles (2-3 nm resolution) the curves are rapidly decaying functions of s essentially determined by the particle shape, which clearly differ.
First applications date back to the late 1930s when the main principles of SAXS were developed in the fundamental work of Guinier following his studies of metallic alloys.
In the first monograph on SAXS by Guinier and Fournet it was already demonstrated that the method yields not only information on the sizes and shapes of particles but also on the internal structure of disordered and partially ordered systems.
A breakthrough in SAXS and SANS experiments came in the 1970s, thanks to the availability of synchrotron radiation and neutron sources, the latter paving the way for contrast variation by solvent exchange of H2O for D2O and specific deuteration methods.
It was realised that scattering studies on solution provide, at a minimal investment of time and effort, useful insights into the structure of non-crystalline biochemical systems.
In the past, only overall particle parameters (e.g. volume, radius of gyration) of the macromolecules were directly determined from the experimental data, whereas the analysis in terms of three-dimensional models was limited to simple geometrical bodies (e.g. ellipsoids, cylinders, etc.)
The 1990s brought a breakthrough in SAXS/SANS data analysis methods, which opened the way for reliable ab initio modelling of macromolecular complexes, including detailed determination of shape and domain structure and application of rigid body refinement techniques.
This progress was accompanied by further advances in instrumentation, allowing sub-ms time resolutions to be achieved on third generation SR sources in the studies of protein and nucleic acid folding.
Small-Angle X-Ray scattering Initiative for EuRope (SAXIER) with the goal to combine SAXS methods with other analytical techniques and create automated software to rapidly analyse large quantities of data.
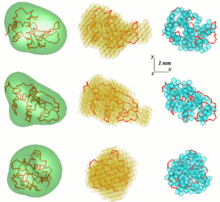
DALAI_GA is an elegant program, which takes a sphere with diameter equal to the maximum particle size Dmax, which is determined from the scattering data, and fills it with beads.
The 'give-n-take' procedure SAXS3D and the program SASMODEL, based on interconnected ellipsoids are ab initio Monte Carlo approaches without limitation in the search space.
[6] An approach that uses an ensemble of Dummy Residues (DRs) and simulated annealing to build a locally "chain-compatible" DR-model inside a sphere of diameter Dmax lets one extract more details from SAXS data.
Depending on the complexity of the object, different approaches are employed for the global search of the optimum configuration of subunits fitting the experimental data.
[6][14][15] Disordered surface amino acids ("loops") are frequently unobserved in NMR and crystallographic studies, and may be left missing in the reported models.
The Dummy Residue approach was extended and the algorithms for adding missing loops or domains were implemented in the program suite CREDO.
Ab initio methods, on the other hand, challenge one of the biggest problems in molecular biology, namely, to predict the folding of a protein "from scratch", using no homologous sequences or structures.
[18] Another example shows how the SAXS data can be combined together with NMR, X-ray crystallography and electron microscopy to reconstruct the quaternary structure of multidomain protein.
IsGISAXS (grazing incidence small angle X-ray scattering) is a software program dedicated to the simulation and analysis of GISAXS from nanostructures.
The particle form factor is calculated within the distorted wave Born approximation (DWBA), starting as an unperturbed state with sharp interfaces or with the actual perpendicular profile of refraction index.