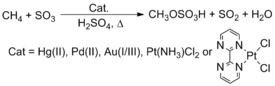Carbon–hydrogen bond activation
In particular, this definition does not require the cleaved C–H bond to initially interact with the transition metal in the reaction mechanism.
[2] In contrast to the organometallic variety, this broadened type of C-H activation is widely employed industrially and in nature.
Other mechanistic possibilities not involving direct C–H bond cleavage by the metal include (i) generation of arylmetal species by electrophilic aromatic substitution mechanism (common for electrophilic Pd, Pt, Au, Hg species), (ii) cleavage of the C–H bond via hydrogen atom abstraction by an O- or N-centered radical, which may then go on to further react and undergo functionalization with or without forming an organometallic intermediate (e.g., Kharasch–Sosnovsky reaction), and (iii) C–H deprotonation at the α-position of a π-system assisted by initial formation of a π-complex with an electrophilic metal to generate a nucleophilic organometallic species (e.g., by cyclopentadienyliron complexes).
R. G. Bergman reported the first transition metal-mediated intermolecular C–H activation of unactivated and completely saturated hydrocarbons by oxidative addition.
The selective activation and functionalization of alkane C–H bonds was reported using a tungsten complex outfitted with pentamethylcyclopentadienyl, nitrosyl, allyl and neopentyl ligands, Cp*W(NO)(η3-allyl)(CH2CMe3).
This transformation was achieved via the thermolysis of Cp*W(NO)(η3-allyl)(CH2CMe3) in pentane at room temperature, resulting in elimination of neopentane by a pseudo-first-order process, generating an undetectable electronically and sterically unsaturated 16-electron intermediate that is coordinated by an η2-butadiene ligand.
Subsequent intermolecular activation of a pentane solvent molecule then yields an 18-electron complex possessing an n-pentyl ligand.
Photoinitiated reactions of transition metal complexes with alkanes serve as a powerful model systems for understanding the cleavage of the strong C-H bond.
However, it is only limited to complexes which have IR-active ligands and is prone to correct assignments on the femtosecond timescale due to underlying vibrational cooling.
[14] Directed-, chelation-assisted-, or "guided" C-H activation involves directing groups that influence regio- and stereochemistry.
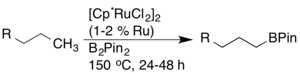
John F. Hartwig reported a highly regioselective arene and alkane borylation catalyzed by a rhodium complex.
Although chemists have failed to develop a commercial process for selective C-H activation of methane, such a reaction is the basis of reverse methanogenesis.
Current technology makes prodigious use of methane by steam reforming to produce syngas, a mixture of carbon monoxide and hydrogen.
This syngas is then used in Fischer-Tropsch reactions to make longer carbon chain products or methanol, one of the most important industrial chemical feedstocks.
Roy A. Periana, for example, reported that complexes containing late transition metals, such as Pt, Pd, Au, and Hg, react with methane (CH4) in H2SO4 to yield methyl bisulfate.
[31] The synthesis of a mescaline analogue employs the rhodium-catalyzed enantioselective annulation of an aryl imine via a C-H activation.