Cladistics
Cladistics (/kləˈdɪstɪks/ klə-DIST-iks; from Ancient Greek κλάδος kládos 'branch')[1] is an approach to biological classification in which organisms are categorized in groups ("clades") based on hypotheses of most recent common ancestry.
The evidence for hypothesized relationships is typically shared derived characteristics (synapomorphies) that are not present in more distant groups and ancestors.
However, from an empirical perspective, common ancestors are inferences based on a cladistic hypothesis of relationships of taxa whose character states can be observed.
Radiation results in the generation of new subclades by bifurcation, but in practice sexual hybridization may blur very closely related groupings.
[6] Branches down to the divergence to the next significant (e.g. extant) sister are considered stem-groupings of the clade, but in principle each level stands on its own, to be assigned a unique name.
Cladistics findings are posing a difficulty for taxonomy, where the rank and (genus-)naming of established groupings may turn out to be inconsistent.
[8] What is now called the cladistic method appeared as early as 1901 with a work by Peter Chalmers Mitchell for birds[9][10] and subsequently by Robert John Tillyard (for insects) in 1921,[11] and W. Zimmermann (for plants) in 1943.
From the time of his original formulation until the end of the 1970s, cladistics competed as an analytical and philosophical approach to systematics with phenetics and so-called evolutionary taxonomy.
Phenetics was championed at this time by the numerical taxonomists Peter Sneath and Robert Sokal, and evolutionary taxonomy by Ernst Mayr.
[17] Originally conceived, if only in essence, by Willi Hennig in a book published in 1950, cladistics did not flourish until its translation into English in 1966 (Lewin 1997).
In the 1990s, the development of effective polymerase chain reaction techniques allowed the application of cladistic methods to biochemical and molecular genetic traits of organisms, vastly expanding the amount of data available for phylogenetics.
At the same time, cladistics rapidly became popular in evolutionary biology, because computers made it possible to process large quantities of data about organisms and their characteristics.
The outcome of a cladistic analysis is a cladogram – a tree-shaped diagram (dendrogram)[18] that is interpreted to represent the best hypothesis of phylogenetic relationships.
Although traditionally such cladograms were generated largely on the basis of morphological characters and originally calculated by hand, genetic sequencing data and computational phylogenetics are now commonly used in phylogenetic analyses, and the parsimony criterion has been abandoned by many phylogeneticists in favor of more "sophisticated" but less parsimonious evolutionary models of character state transformation.
Cladists contend that these models are unjustified because there is no evidence that they recover more "true" or "correct" results from actual empirical data sets [19] Every cladogram is based on a particular dataset analyzed with a particular method.
Cladograms that are supported by a large number and variety of different kinds of characters are viewed as more robust than those based on more limited evidence.
Decisions as to whether particular character states are homologous, a precondition of their being synapomorphies, have been challenged as involving circular reasoning and subjective judgements.
Horizontal gene transfer is the mobility of genetic info between different organisms that can have immediate or delayed effects for the reciprocal host.
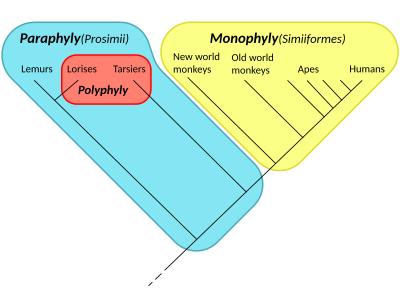
Furthermore, established names are discarded in cladistics, or alternatively carry connotations which may no longer hold, such as when additional groups are found to have emerged in them.
Mythological phylogenies constructed with mythemes clearly support low horizontal transmissions (borrowings), historical (sometimes Palaeolithic) diffusions and punctuated evolution.
This is similar to the traditional comparative method of historical linguistics, but is more explicit in its use of parsimony and allows much faster analysis of large datasets (computational phylogenetics).