Cystic fibrosis
[16]: Clinical Manifestations The primary cause of morbidity and death in people with cystic fibrosis is progressive lung disease, which eventually leads to respiratory failure.
Approximately 15%-20% of newborns diagnosed with CF experience intestinal blockage (meconium ileus), and other digestive issues may arise due to mucus accumulation in the pancreas.
[29] The thick mucus seen in the lungs has a counterpart in thickened secretions from the pancreas, an organ responsible for providing digestive juices that help break down food.
These secretions block the exocrine movement of the digestive enzymes into the duodenum and result in irreversible damage to the pancreas, often with painful inflammation (pancreatitis).
[20]: 1254 Despite this, idiopathic chronic pancreatitis can occur in a subset of pancreas-sufficient individuals with CF, and is associated with recurrent abdominal pain and life-threatening complications.
Polymorphisms in one or both mannan-binding lectin alleles that result in lower circulating levels of the protein are associated with a threefold higher risk of end-stage lung disease, as well as an increased burden of chronic bacterial infections.
[52] The CFTR gene regulates the transport of salts and water through cell membranes, providing instructions for creating a pathway that allows the passage of chloride ions.
[63] As cilia cannot effectively move in a thick, viscous environment, mucociliary clearance is deficient and a buildup of mucus occurs, clogging small airways.
[64] The accumulation of more viscous, nutrient-rich mucus in the lungs allows bacteria to hide from the body's immune system, causing repeated respiratory infections.
[65] Scientific evidence suggests the interleukin 17 pathway plays a key role in resistance and modulation of the inflammatory response during P. aeruginosa infection in CF.
[67] In particular, interleukin 17-mediated immunity plays a double-edged activity during chronic airways infection; on one side, it contributes to the control of P. aeruginosa burden, while on the other, it propagates exacerbated pulmonary neutrophilia and tissue remodeling.
[88] Treatment for CF is diverse, tailored to different symptoms, and includes various devices, inhalation medications to alleviate respiratory difficulties, oral enzyme supplements to address exocrine pancreatic insufficiency, and, in some cases, surgical interventions for conditions such as meconium ileus.
[citation needed] Because of the wide variation in disease symptoms, treatment typically occurs at specialist multidisciplinary centers and is tailored to the individual.
[93] The most consistent aspect of therapy in CF is limiting and treating the lung damage caused by thick mucus and infection, with the goal of maintaining quality of life.
[citation needed] Antibiotics are absolutely necessary whenever pneumonia is suspected or a noticeable decline in lung function is seen, and are usually chosen based on the results of a sputum analysis and the person's past response.
Inhaled therapy with antibiotics such as tobramycin, colistin, and aztreonam is often given for months at a time to improve lung function by impeding the growth of colonized bacteria.
[106] Methicillin-resistant Staphylococcus aureus (MRSA) infections can be dangerous for people with cystic fibrosis and can worsen lung damage leading to more rapid decline.
[143] However, the authors noted that "non-invasive ventilation may be a useful adjunct to other airway clearance techniques, particularly in people with cystic fibrosis who have difficulty expectorating sputum".
Treatment of pancreatic insufficiency by replacement of missing digestive enzymes allows the duodenum to properly absorb nutrients and vitamins that would otherwise be lost in the feces.
[157] Bisphosphonates taken by mouth or intravenously can be used to improve the bone mineral density in people with cystic fibrosis, but there are no proof that this reduces fractures or increases survival rates.
In 2020 the median predicted life expectancy was around 59 years, although there are uncertainties in the estimates due to the low number of annual deaths for persons with cystic fibrosis.
[citation needed] According to Schmitz and Goldbeck (2006), CF significantly increases emotional stress on both the individual and the family, "and the necessary time-consuming daily treatment routine may have further negative effects on quality of life".
[176] However, Havermans and colleagues (2006) have established that young outpatients with CF who have participated in the Cystic Fibrosis Questionnaire-Revised "rated some quality of life domains higher than did their parents".
[178] No definitive cure for CF is known, but diverse medications are used, such as mucolytics, bronchodilators, steroids, and antibiotics, that have the purpose of loosening mucus, expanding airways, decreasing inflammation, and fighting lung infections, respectively.
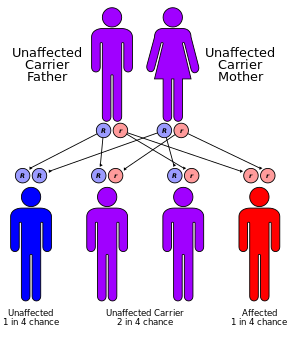
Resistance to the following have all been proposed as possible sources of heterozygote advantage: CF is supposed to have appeared about 3,000 BC because of migration of peoples, gene mutations, and new conditions in nourishment.
Indeed, literature from Germany and Switzerland in the 18th century warned "Wehe dem Kind, das beim Kuß auf die Stirn salzig schmeckt, es ist verhext und muss bald sterben" ("Woe to the child who tastes salty from a kiss on the forehead, for he is bewitched and soon must die"), recognizing the association between the salt loss in CF and illness.
[215][217] People with CF may be listed in a disease registry that allows researchers and doctors to track health results and identify candidates for clinical trials.
As such, the bacteriophage therapy makes is a promising alternative for treating infections caused by multidrug-resistant bacteria, such as Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa in CF patients, which are often protected by biofilms and thus resistant to conventional antibiotics.
In some cases, they can cause the cell to overcome a premature stop codon by inserting a random amino acid, thereby allowing expression of a full-length protein.
[236] Due to this, it has been suggested that the direct alteration of CF microbial community composition and metabolic function would provide an alternative to traditional antibiotic therapies.


Green = Pseudomonas aeruginosa
Brown = Staphylococcus aureus
Blue = Haemophilus influenzae
Red = Burkholderia cepacia complex