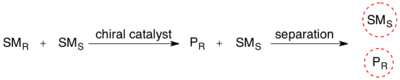
Kinetic resolution
The enantiomeric excess (ee) of the unreacted starting material continually rises as more product is formed, reaching 100% just before full completion of the reaction.
Kinetic resolution by synthetic means was first reported by Marckwald and McKenzie in 1899 in the esterification of racemic mandelic acid with optically active (−)-menthol.
[4][5] Kinetic resolution is a possible method for irreversibly differentiating a pair of enantiomers due to (potentially) different activation energies.
[6] The selectivity can also be expressed in terms of ee of the recovered starting material and conversion (c), if first-order kinetics (in substrate) are assumed.
As a result, a common approach is to measure and report only yields and ee's, as the formula for krel only applies to an idealized kinetic resolution.
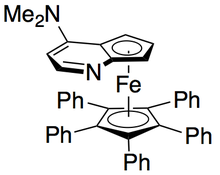
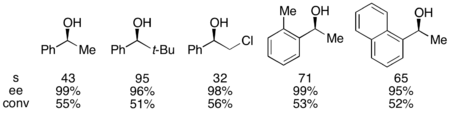
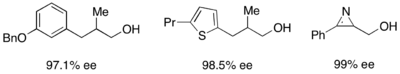
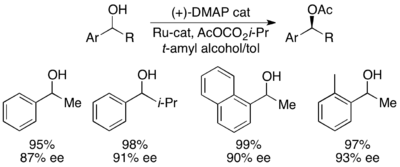
Gregory Fu and colleagues have developed a methodology utilizing a chiral DMAP analogue to achieve excellent kinetic resolution of secondary alcohols.
[7] Initial studies utilizing ether as a solvent, low catalyst loadings (2 mol %), acetic anhydride as the acylating agent, and triethylamine at room temperature gave selectivities ranging from 14-52, corresponding to ee's of the recovered alcohol product as high as 99.2%.
The limitations of this method include the requirement of an unsaturated functionality, such as carbonyl or alkenes, at the remote alkynyl position.
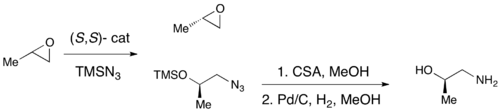
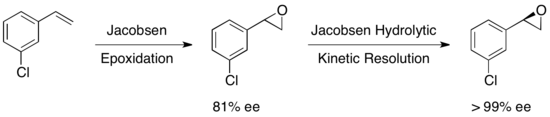
In 1996, Jacobsen and coworkers developed a methodology for the kinetic resolution of epoxides via nucleophilic ring-opening with attack by an azide anion.
In 1997, Jacobsen's group published a methodology which improved upon their earlier work, allowing for the use of water as the nucleophile in the epoxide opening.
The advantage of this approach is the ability to reduce the amount of hydrolytic cleavage necessary to achieve high enantioselectivity, allowing for overall yields up to approximately 90%, based on the olefin.
Quite recently, D. A. Devalankar et al. reported an elegant protocol involving a two-stereocentered Co-catalyzed HKR of racemic terminal epoxides bearing adjacent C–C binding substituents.
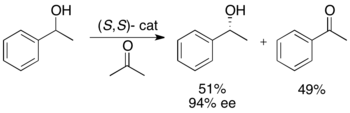
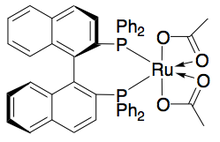
[24] Ryōji Noyori and colleagues have developed a methodology for the kinetic resolution of benzylic and allylic secondary alcohols via transfer hydrogenation.
In the example illustrated below, exposure of 1-phenylethanol to the (S,S) enantiomer of the catalyst in the presence of acetone results in a 51% yield of 94% ee (R)-1-phenylethanol, along with 49% acetophenone and isopropanol as a byproduct.
[25] This methodology is essentially the reverse of Noyori's asymmetric transfer hydrogenation of ketones,[26] which yield enantioenriched alcohols via reduction.
Thus, the kinetic resolution would only be carried out in an instance for which the racemic alcohol was at least one half the price of the ketone or significantly easier to access.
However, slight structural changes in the substrate, such as increasing the inter-alkene distance to 1,7, can sometimes necessitate the use of a different catalyst, reducing the efficacy of this method.
On a commercial scale, Degussa's methodology employing acylases is capable of resolving numerous natural and unnatural amino acids.
[33] The use of isopropenyl acetate as the acylating agent results in acetone as the byproduct, which is effectively removed from the reaction using molecular sieves.
This is equivalent to writing, for kR>kS, A number of excellent reviews have been published, most recently in 2008, detailing the theory and practical applications of DKR.
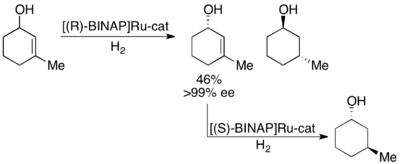
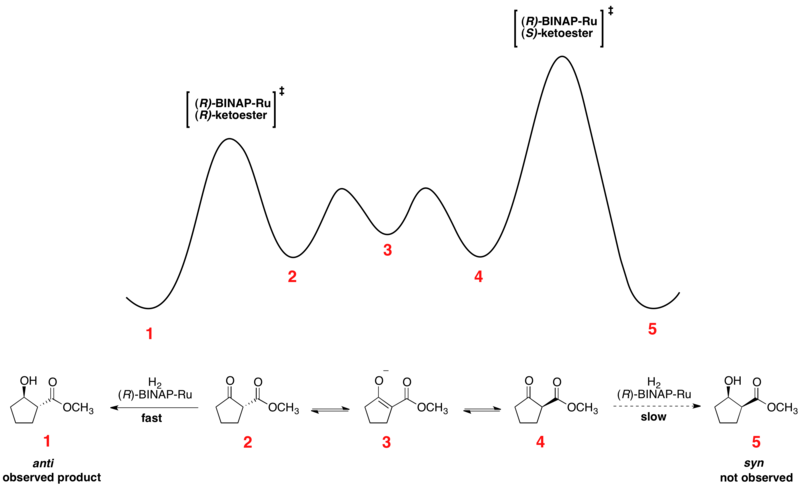
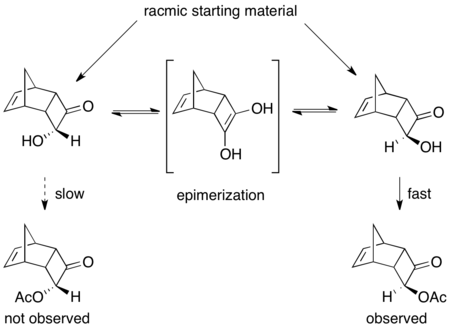
The enantiomeric β-ketoesters can undergo epimerization, and the choice of chiral catalyst, typically of the form Ru[(R)-BINAP]X2, where X is a halogen, leads to one of the enantiomers reacting preferentially faster.
[41][42] As can be seen, the epimerization intermediate is lower in free energy than the transition states for hydrogenation, resulting in rapid racemization and high yields of a single enantiomer of the product.
Solvent choice appears to have a major influence on the diastereoselectivity, as dichloromethane and methanol both show effectiveness for certain substrates.
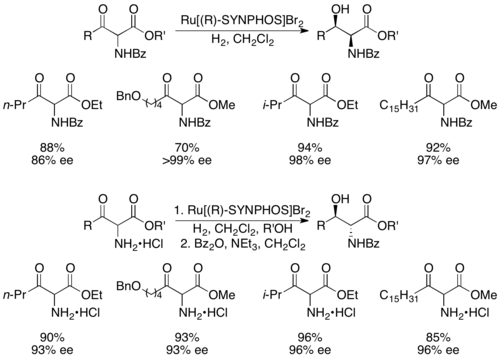
Genêt and coworkers developed SYNPHOS, a BINAP analogue which forms ruthenium complexes, which perform highly selective asymmetric hydrogenations.
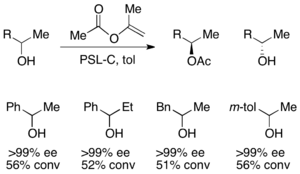
[46] A prime example using PSL effectively resolves racemic acyloins in the presence of triethylamine and vinyl acetate as the acylating agent.
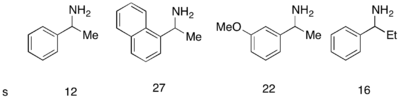
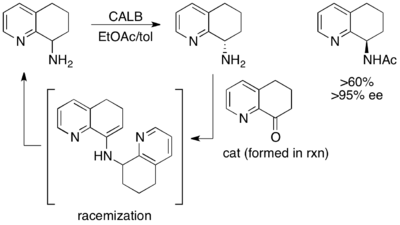
[48] Here, the unreacted starting material racemizes in situ via a dimeric enamine, resulting in a recovery of greater than 50% yield of the enantiopure acylated amine product.
There have been a number of reported procedures which take advantage of a chemical reagent/catalyst to perform racemization of the starting material and an enzyme to selectively react with one enantiomer, called chemoenzymatic dynamic kinetic resolutions.
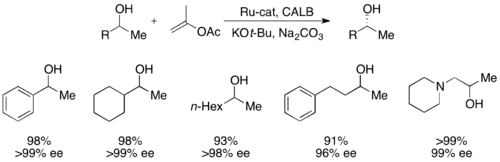
[50] More recently, secondary alcohols have been resolved by Bäckvall with yields up to 99% and ee's up to >99% utilizing CALB and a ruthenium racemization complex.
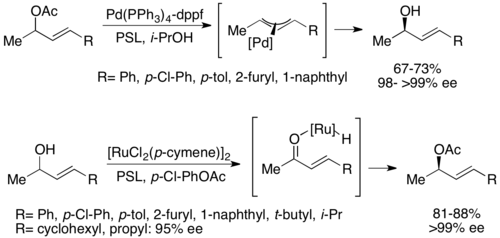
[51] A second type of chemoenzymatic dynamic kinetic resolution involves a π-allyl complex from an allylic acetate with palladium.
[53] In parallel kinetic resolution (PKR), a racemic mixture reacts to form two non-enantiomeric products, often through completely different reaction pathways.