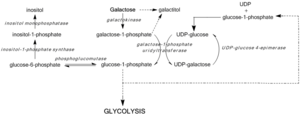
Galactose epimerase deficiency
Symptoms of congenital Type III Galactosemia are apparent from birth, but vary in severity depending on whether the peripheral or generalized disease form is present.
The recent development of a Drosophila melanogaster GALE mutant exhibiting galactosemic symptoms may yield a promising future animal model.
[6] Functional analysis of these mutant GALE isoforms suggests that reduced catalytic efficiency and increased likelihood of proteolytic digestion act causatively in Type III galactosemia.
High galactose-1-phosphate levels have been shown to interfere with phosphoglucomutase,[7] glycogen phosphorylase,[8] UDP-glycopyrophosphorylase,[9] activity in bacterial models and in vitro, yet in vivo mechanisms toxicity have yet to be confirmed.
[citation needed] Individuals presenting with Type III galactosemia must consume a lactose- and galactose-restricted diet devoid of dairy products and mucilaginous plants.