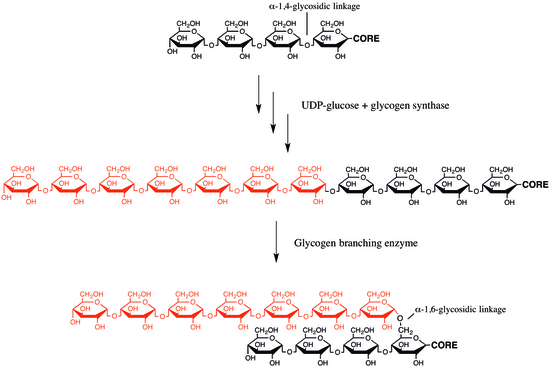
Glycogen branching enzyme
Branching of the chains is essential to increase the solubility of the glycogen molecule and, consequently, in reducing the osmotic pressure within cells.
[5][7][8][9] Through Southern blot analysis of DNA derived from human/rodent somatic cell hybrids, GBE1 has been identified as an autosomal gene located on the short arm of chromosome 3 at position 12.3.
[10] Additionally, the coding sequence was found to comprise 2,106 base pairs and encode a 702-amino acid long GBE.
Additionally, there are striking differences between the loops connecting elements of the secondary structure among these various α-amylase members, especially around the active site.
[11] Glycogen binding enzymes in other organisms have also been crystallized and structurally determined, demonstrating both similarity and variation to GBE found in Escherichia coli.
[9] This disease is characterized by a severe depletion or complete absence of GBE, resulting in the accumulation of abnormally structured glycogen, known as polyglucosan bodies.
[9] Glycogen buildup leads to increased osmotic pressure resulting in cellular swelling and death.
[9] The tissues most affected by this disease are the liver, heart, and neuromuscular system, areas with the greatest levels of glycogen accumulation.
Specifically, some disease characteristics are gait difficulties from mixed upper and lower motor neuron involvement sensory loss in lower extremities, and neurogenic bladder, a problem in which a person lacks bladder control due to a brain, spinal cord, or nerve condition.