Hyperconjugation
In organic chemistry, hyperconjugation (σ-conjugation or no-bond resonance) refers to the delocalization of electrons with the participation of bonds of primarily σ-character.
More controversially, hyperconjugation is proposed by quantum mechanical modeling to be a better explanation for the preference of the staggered conformation rather than the old textbook notion of steric hindrance.
Their work, first published in 1937, was intended as a preliminary progress report of thermochemical studies of energy changes during addition reactions of various unsaturated and cyclic compounds.
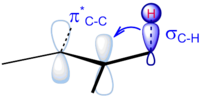
[12] The key interaction is believed to be the donation of electron density from the neighboring C–H σ bond into the π* antibonding orbital of the alkene (σC–H→π*).
The effect is almost an order of magnitude weaker than the case of alkyl substitution on carbocations (σC–H→pC), since an unfilled p orbital is lower in energy, and, therefore, better energetically matched to a σ bond.
One set of experiments by Kistiakowsky involved collected heats of hydrogenation data during gas-phase reactions of a range of compounds that contained one alkene unit.
These experiments revealed an important result; when n=0, there is an effect of conjugation to the molecule where the ΔH value is lowered by 3.5 kcal.
Kistiakowsky also investigated open chain systems, where the largest value of heat liberated was found to be during the addition to a molecule in the 1,4-position.
[14] Another group led by Houk[15] suggested the methods employed by Rogers and Kistiakowsky was inappropriate, because that comparisons of heats of hydrogenation evaluate not only conjugation effects but also other structural and electronic differences.
A relatively recent work (2006) by Fernández and Frenking (2006) summarized the trends in hyperconjugation among various groups of acyclic molecules, using energy decomposition analysis or EDA.
Fernández and Frenking reported that the methyl, hydroxyl, and amino substituents resulted in a decrease in ΔEpi from the parent 2-propenal.
Wilson had proven that the energy barrier between any pair of eclipsed and staggered conformations is approximately 3 kcal/mol, and the generally accepted rationale for this was the unfavorable steric interactions between hydrogen atoms.
The analysis of the curves determined that the staggered conformation had no connection to the amount of electrostatic repulsions within the molecule.
These results demonstrate that Coulombic forces do not explain the favored staggered conformations, despite the fact that central bond stretching decreases electrostatic interactions.