Lewis acid catalysis
The complexation has partial charge-transfer character and makes the lone-pair donor effectively more electronegative, activating the substrate toward nucleophilic attack, heterolytic bond cleavage, or cycloaddition with 1,3-dienes and 1,3-dipoles.
Over the years, versatile catalysts bearing ligands designed for specific applications have facilitated improvement in both reactivity and selectivity of Lewis acid-catalyzed reactions.
Studying the product ratio in a bicyclic system, Denmark and colleagues showed that both mechanisms could be operative depending on the denticity of the Lewis acid and the identity of the R' group.
[3] In Diels-Alder and 1,3-dipolar cycloaddition reactions, Lewis acids lower the LUMO energy of the dienophile or dipolarphile, respectively, making it more reactive toward the diene or the dipole.
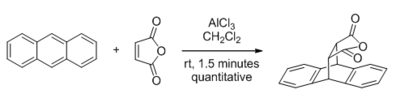
The first major discovery in this area was in 1960, when Yates and Eaton reported the significant acceleration of the Diels-Alder reaction by AlCl3 when maleic anhydride is the dienophile.
[4] Early theoretical studies that depended on frontier orbital analysis established that Lewis acid catalysis operates via lowering of the dienophile's LUMO energy,.
In addition, the Lewis acid catalyst also increases the asynchronicity of the Diels-Alder reaction, making the occupied π-orbital located on the C=C double bond of the dienophile asymmetric.
This is in contrast with the rigid Zimmerman-Traxler cyclic transition state that has been widely accepted for the aldol reaction with lithium, boron, and titanium enolates.
The Lewis acid catalyst plays a role in stereoselectivity when the aldehyde can chelate onto the metal center and form a rigid cyclic intermediate.
In a seminal review in the early 1990s, Mikami and colleagues[29] proposed a late, chair-like transition state, which could rationalize many observed stereochemical results, including the role of steric bulk in diastereoselectivity:[30] More recently, however, the same group carried out HF/6-31G* calculations on tin or aluminum Lewis acid-catalyzed ene reactions.
[34] The Diels-Alder reaction catalyzed or promoted by Lewis acids is a powerful and widely used method in natural product synthesis to attain scaffold complexity in a single step with stereochemical control.
Over the years, a small number of chiral ligand scaffolds have stood out as having "privileged" catalytic properties suitable for a wide range of applications, often of unrelated mechanisms.
Current research efforts in asymmetric Lewis acid catalysis mostly utilize or modify those ligands rather than create new scaffolds de novo.
[52] On the other hand, two-point binding on a Lewis acid bearing the meridionally tridentate PyBOX ligand would result in a square pyramidal complex.
[53] Developed by Noyori, BINAP (2,2'-diphenylphosphino-1,1'-binaphthyl) is a family of chiral diphosphine ligands featuring two triarylphosphine moieties installed on a binaphthalene backbone.
For example, in the palladium-catalyzed enantioselective Diels-Alder reaction shown below, the dienophile is thought to coordinate the metal center at the equatorial sites.
[56] A very similar model was used to rationalize the outcome of a nickel-catalyzed asymmetric enolate alkylation reaction, where the substrate also bears an auxiliary that allows it to chelate onto the metal.
[57] On the other hand, a copper(I)-catalyzed hetero-ene reaction is thought to proceed through a tetrahedral intermediate,[58] offering an alternative mode of stereoinduction by changing the metal center.
The broad application of titanium TADDOLate catalysts towards carbonyl additions and cycloadditions has been introduced by Seebach and coworkers, and has been thoroughly summarized in a seminal review, in which a working stereoinduction model that agreed with the observed selectivity in a wide variety of reactions was put forth, despite the lack of a clear picture of the mechanism.