Neuromuscular junction
Genetic disorders, such as Congenital myasthenic syndrome, can arise from mutated structural proteins that comprise the neuromuscular junction, whereas autoimmune diseases, such as myasthenia gravis, occur when antibodies are produced against nicotinic acetylcholine receptors on the sarcolemma.
At the neuromuscular junction, presynaptic motor axons terminate 30 nanometers from the cell membrane or sarcolemma of a muscle fiber.
The sarcolemma at the junction has invaginations called postjunctional folds, which increase its surface area facing the synaptic cleft.
The 30 nanometer cleft between nerve ending and endplate contains a meshwork of acetylcholinesterase (AChE) at a density of 2,600 enzyme molecules/μm2, held in place by the structural proteins dystrophin and rapsyn.
[4] About once every second in a resting junction randomly one of the synaptic vesicles fuses with the presynaptic neuron's cell membrane in a process mediated by SNARE proteins.
The acetylcholine quantum diffuses through the acetylcholinesterase meshwork, where the high local transmitter concentration occupies all of the binding sites on the enzyme in its path.
This influx of Ca2+ causes several hundred neurotransmitter-containing vesicles to fuse with the presynaptic neuron's cell membrane through SNARE proteins to release their acetylcholine quanta by exocytosis.
The EPP is accomplished when ACh binds the nicotinic acetylcholine receptors (nAChR) at the motor end plate, and causes an influx of sodium ions.
The transmission from nerve to muscle is so rapid because each quantum of acetylcholine reaches the endplate in millimolar concentrations, high enough to combine with a receptor with a low affinity, which then swiftly releases the bound transmitter.
[citation needed] Acetylcholine is a neurotransmitter synthesized from dietary choline and acetyl-CoA (ACoA), and is involved in the stimulation of muscle tissue in vertebrates as well as in some invertebrate animals.
[4] AChRs at the skeletal neuromuscular junction form heteropentamers composed of two α, one β, one ɛ, and one δ subunits.
[5] The presence of the inactive, intermediate receptor structure with a single-bound ligand keeps ACh in the synapse that might otherwise be lost by cholinesterase hydrolysis or diffusion.
[11] The development of the neuromuscular junction requires signaling from both the motor neuron's terminal and the muscle cell's central region.
During development, muscle cells produce acetylcholine receptors (AChRs) and express them in the central regions in a process called prepatterning.
[16] In this model presynaptic motor neurons are activated by optogenetics and in response synaptically connected muscle fibers twitch upon light stimulation.
José del Castillo and Bernard Katz used ionophoresis to determine the location and density of nicotinic acetylcholine receptors (nAChRs) at the neuromuscular junction.
The intracellular microelectrode monitored the amplitude of the depolarization of the motor endplate in response to ACh binding to nicotinic (ionotropic) receptors.
Katz and del Castillo showed that the amplitude of the depolarization (excitatory postsynaptic potential) depended on the proximity of the micropipette releasing the ACh ions to the endplate.
α-Bungarotoxin is a toxin found in the snake species Bungarus multicinctus that acts as an ACh antagonist and binds to AChRs irreversibly.
The accumulation of ACh within the synaptic cleft causes muscle cells to be perpetually contracted, leading to severe complications such as paralysis and death within minutes of exposure.
[4] This toxin crosses into the nerve terminal through the process of endocytosis and subsequently cleaves SNARE proteins, preventing the ACh vesicles from fusing with the intracellular membrane.
Latrotoxin (α-Latrotoxin) found in venom of widow spiders also affects the neuromuscular junction by causing the release of acetylcholine from the presynaptic cell.
Those that inhibit neurotransmitter release create a neuromuscular blockade that prevents signaling molecules from reaching their postsynaptic target receptors.
After one hour of inoculation of these toxins, including notexin and taipoxin, many of the affected nerve terminals show signs of irreversible physical damage, leaving them devoid of any synaptic vesicles.
[18] Any disorder that compromises the synaptic transmission between a motor neuron and a muscle cell is categorized under the umbrella term of neuromuscular diseases.
[22] Proximal muscle weakness is a product of pathogenic autoantibodies directed against P/Q-type voltage-gated calcium channels, which in turn leads to a reduction of acetylcholine release from motor nerve terminals on the presynaptic cell.
Specifically, these syndromes are diseases incurred due to mutations, typically recessive, in 1 of at least 10 genes that affect presynaptic, synaptic, and postsynaptic proteins in the neuromuscular junction.
3,4-Diaminopyridine, the first-line treatment for LEMS, is under development as an orphan drug for CMS[29] in the US, and available to eligible patients under an expanded access program at no cost.

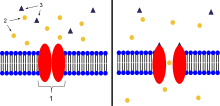
- Ion channel linked receptor
- Ions
- Ligand (such as acetylcholine )
