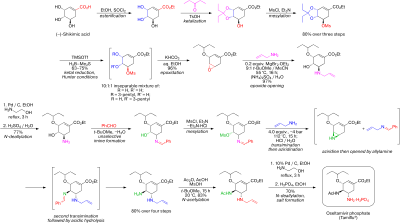
Oseltamivir total synthesis
Reductive opening of the ketal under modified Hunter conditions[8] in dichloromethane yields an inseparable mixture of isomeric mesylates.
The water-immiscible solvents methyl tert-butyl ether and acetonitrile are used to simplify the workup procedure, which involved stirring with 1 M aqueous ammonium sulfate.
The aminoalcohol was converted directly to the corresponding allyl-diamine in an interesting cascade sequence that commences with the unselective imination of benzaldehyde with azeotropic water removal in methyl tert-butyl ether.
Mesylation, followed by removal of the solid byproduct triethylamine hydrochloride, results in an intermediate that was poised to undergo aziridination upon transimination with another equivalent of allylamine.
Selective acylation with acetic anhydride (under buffered conditions, the 5-amino group is protonated owing to a considerable difference in pKa, 4.2 vs 7.9, preventing acetylation) yields the desired N-acetylated product in crystalline form upon extractive workup.
Butadiene 1 reacts in an asymmetric Diels-Alder reaction with the esterification product of acrylic acid and 2,2,2-trifluoroethanol 2 catalysed by the CBS catalyst.
The ester 3 is converted into an amide in 4 by reaction with ammonia and the next step to lactam 5 is an iodolactamization with iodine initiated by trimethylsilyl triflate.
The newly formed double bond is functionalized with N-bromoacetamide 10 catalyzed with tin(IV) bromide with complete control of stereochemistry.
Cyanophosphorylation with diethyl phosphorocyanidate (DEPC) modifies the ketone group to the cyanophosphate 10 paving the way for an intramolecular allylic rearrangement to unstable β-allyl phosphate 11 (toluene, sealed tube) which is hydrolyzed to alcohol 12 with ammonium chloride.
The asymmetric Diels-Alder reaction with acrolein 3 is carried out with the McMillan catalyst to the aldehyde 4 as the endo isomer which is oxidized to the carboxylic acid 5 with sodium chlorite, monopotassium phosphate and 2-methyl-2-butene.
Keeping cost, yield, and number of synthetic steps in mind, an enantioselective total synthesis of (1) was accomplished through three one-pot operations.
[14] In the second one-pot operation, trifluoroacetic acid is employed first to deprotect the tert-butyl ester of (6); any excess reagent was removed via evaporation.
[14] The final one-pot operation begins with a Curtius Rearrangement of acyl azide (7) to produce an isocyanate functional group at room temperature.
This domino Curtius rearrangement and amide formation occurs in the absence of heat, which is extremely beneficial for reducing any possible hazard.