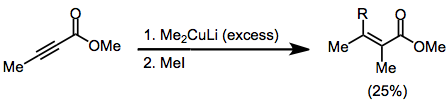Vicinal difunctionalization
Vicinal difunctionalization refers to a chemical reaction involving transformations at two adjacent centers (most commonly carbons).
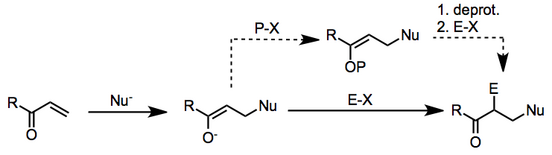
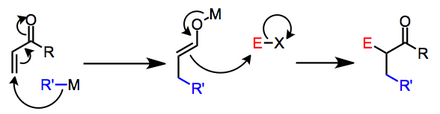
This transformation can be accomplished in α,β-unsaturated carbonyl compounds via the conjugate addition of a nucleophile to the β-position followed by trapping of the resulting enolate with an electrophile at the α-position.
[1] Vicinal difunctionalization reactions, most generally, lead to new bonds at two adjacent carbon atoms.
The mechanism proceeds in two stages: β-nucleophilic addition to the unsaturated carbonyl compound, followed by electrophilic substitution at the α-carbon of the resulting enolate.
[2] Research has shown that the second step may even proceed via single-electron transfers when the reduction potential of the electrophile is low.
The two steps may be carried out as distinct experimental operations if the initially formed enolate is protected after β-addition.
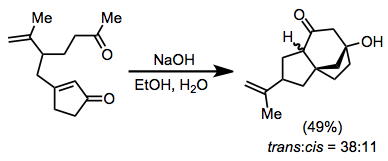
On the basis of steric approach control, the new α-substituent is predicted to be trans to the new β-substituent, and this is observed in a number of cases.
To prevent simple Michael addition (which culminates in protonation of the enolate intermediate), trapping by the electrophile must be intramolecular.
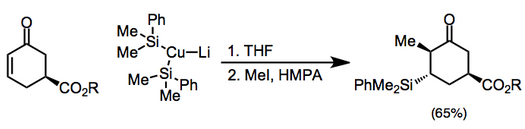
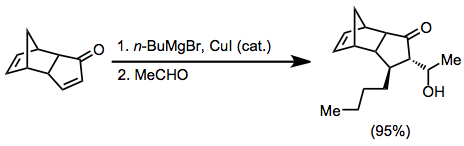
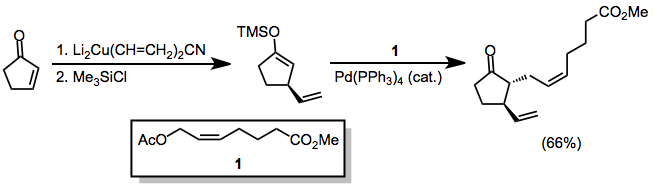
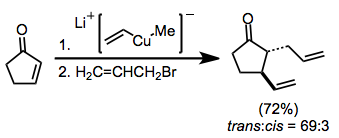
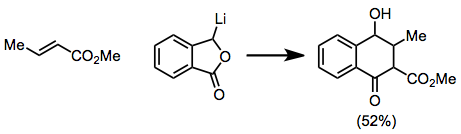
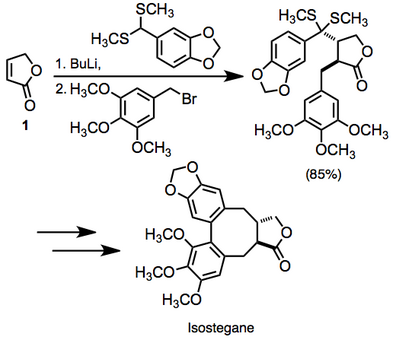
Cyclic α,β-unsaturated ketones are the most commonly employed substrates for vicinal difunctionalization.
[12][13] (9)A large number of examples of vicinal difunctionalization of unsaturated carbonyl compounds exist in the literature.
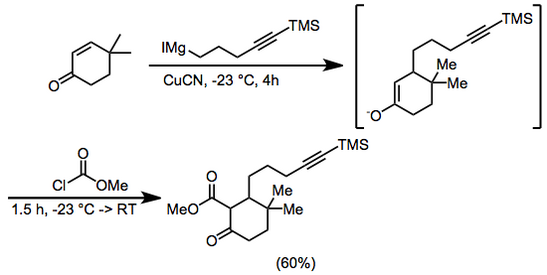
Hydrochloric acid (100 mL, 2.0 M) then was added and the organic phase separated and dried with magnesium sulfate.
The solvent was removed and the residue chromatographed on silica gel using 5% diethyl ether–petroleum ether to give methyl 3,3-dimethyl-6-oxo-2-[5-(trimethylsilyl)-4-pentynyl]cyclohexanecarboxylate, 9.66 g (60%).