Viral quasispecies
Quasispecies result from high mutation rates as mutants arise continually and change in relative frequency as viral replication and selection proceeds.
[4] The term quasispecies was adopted from a theory of the origin of life in which primitive replicons consisted of mutant distributions, as found experimentally with present-day RNA viruses within their host.
However, standard clonal analyses and deep sequencing methodologies have confirmed the presence of myriads of mutant genomes in viral populations, and their participation in adaptive processes.
[1] Quasispecies theory was developed in the 1970s by Manfred Eigen and Peter Schuster to explain self-organization and adaptability of primitive replicons (a term used to refer to any replicating entity), as an ingredient of hypercyclic organizations that link genotypic and phenotypic information, as an essential step in the origin of life.
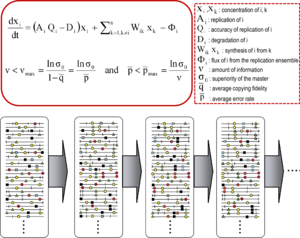
[11][9] The theory portrayed early replicon populations as organized mutant spectra dominated by a master sequence, the one endowed with the highest fitness (replicative capacity) in the distribution.
[1] The existence of a mutant spectrum was experimentally evidenced first by clonal analyses of RNA bacteriophage Qβ populations whose replication had been initiated by a single virus particle.
John Holland and colleagues were the first to recognize that a rapidly evolving RNA world inserted in a DNA-based biosphere had multiple evolutionary and medical implications.
[23] When put in the context of present-day knowledge, we realize that these observations on phenotypic changes were the tip of the iceberg of an extremely complex reality of viral populations.
[10][24][25] The first mathematical formulation of quasispecies was deterministic; it assumed steady state mutant distributions in genetic equilibrium without perturbations derived from modifications of the environment or population size.
[27] Research on quasispecies has proceeded through several theoretical and experimental avenues that include continuing studies on evolutionary optimization and the origin of life, RNA-RNA interactions and replicator networks, the error threshold in variable fitness landscapes, consideration of chemical mutagenesis and proofreading mechanisms, evolution of tumor cells, bacterial populations or stem cells, chromosomal instability, drug resistance, and conformation distributions in prions (a class of proteins with conformation-dependent pathogenic potential; in this case the quasispecies is defined by a distribution of conformations).
[30] Also, postreplicative-repair pathways, abundant to correct genetic lesions in replicating cellular DNA, appear as ineffective for double-stranded RNA or RNA-DNA hybrids.
Quasispecies dynamics will operate in any viral or cellular system in which due to high mutation rates (as a result of low fidelity nucleic acid polymerases or environmental alterations) mutant spectra are rapidly generated.
[22][10][23][24][25][42][43][44] Adaptability of RNA viruses is linked to parameters that facilitate exploration of sequence space: genome size (1.8 to 33 Kb), population size (variable but that can attain an impressive 1012 individual genomes in an infected host at a given time), replication rate, mutation rate, fecundity (yield of viral particles per cell), and number of mutations required for a phenotypic change (surprisingly low for several relevant traits[58]).
In foot-and-mouth disease virus (FMDV) such a design led to a remarkable phenotypic diversification into subpopulations of colonizers and competitors, that modulated virulence of the mutant ensemble.
[15][20][10][43][44][58][60][64] The points summarized in previous sections fully justifies addressing analytical tools towards the mutant spectrum rather than ignoring it or considering its presence a side issue.
Use of consensus sequences to describe the genome of a virus isolate, despite being warranted by the difficulties of conveying the information recapitulated in a mutant spectrum, blurs and enfeebles biological interpretations.
This was illustrated with a FMDV quasispecies that was reconstructed in the laboratory with multiple antigenic variants (each at low frequency) that belonged to two different categories, and shared resistance to the same monoclonal antibody.
[71] In the experiments designed to identify memory in viral quasispecies, members of the mutant spectrum increased in frequency as a consequence of their replication during a selection event that drove them towards dominance.
The two alternative fates are dependent on several factors, one being the surrounding mutant spectrum in those steps of the infectious cycle in which an effective competition among variants is established, for example within replication complexes.
[96][97] This design served to verify experimentally the operation of Müller’s ratchet, or fitness decrease by the irreversible incorporation of mutations in asexual organisms in absence of compensatory mechanisms.
It will be interesting to investigate whether focused adaptation of other viruses to a specific environment may also entail a broadening of diversity, with many phenotypic variants attaining similar fitness levels.
Evolution of viruses in nature and as disease agents can be viewed as succession of mutant spectrum alterations, subjected to expansions and reductions of population size in a continuous interplay of positive and negative selection and random drift.
While short-term (for example, intra-host) evolution is observable and measurable, viruses may appear to be relatively static in the long term for decades (as seen with antigenic variants of FMDV [102]) or longer.
Expressed simply, the direct danger for vaccination and treatment is that the virus can escape through selection of mutants resistant to vaccine-triggered defense components or to the externally administered inhibitors.
[106] Vaccines exposing multiple epitopes and combination therapies follow the same strategy whose aim is to limit possible escape routes to viral quasispecies in the face of the suppressive constraint.
[10][28][44][58][62][83][107][108] Application of lethal mutagenesis as an antiviral strategy deserves attention in the context of the present article because its origins lie in quasispecies theory, in the form of the error threshold relationship.
[83] The term lethal mutagenesis was coined by Lawerence Loeb and colleagues,[107] and it is now widely used to describe the antiviral activity of base and nucleoside analogues that increase the viral mutation rate.
[109][108] Interestingly, some antiviral agents licensed for human use, initially thought to act only as inhibitors of viral replication, may actually exert their antviral activity against some RNA viruses at least partially by lethal mutagenesis.
[108] Defense mechanisms based on genome modification of invading genetic parasites such as editing cellular activities that are recruited as part of the innate immune response (ADAR, APOBEC, RIP, etc.
[3] Over the long-term, a flatter fitness profile might better allow a quasispecies to exploit changes in selection pressure, analogous to the way sexual organisms use recombination to preserve diversity in a population.