Electrophilic aromatic directing groups
[1][2] As a result of these electronic effects, an aromatic ring to which such a group is attached is more likely to participate in electrophilic substitution reaction.
The EWG removes electron density from a π system, making it less reactive in this type of reaction,[2][3] and therefore called deactivating groups.
EDGs and EWGs also determine the positions (relative to themselves) on the aromatic ring where substitution reactions are most likely to take place.
[4] Electron donating groups are typically divided into three levels of activating ability (The "extreme" category can be seen as "strong".)
Weakly deactivating groups direct electrophiles to attack the benzene molecule at the ortho- and para- positions, while strongly and moderately deactivating groups direct attacks to the meta- position.
The partial rate factor of electrophilic aromatic substitution on fluorobenzene is often larger than one at the para position, making it an activating group.
Although the full electronic structure of an arene can only be computed using quantum mechanics, the directing effects of different substituents can often be guessed through analysis of resonance diagrams.
Specifically, any formal negative or positive charges in minor resonance contributors (ones in accord with the natural polarization but not necessarily obeying the octet rule) reflect locations having a larger or smaller density of charge in the molecular orbital for a bond most likely to break.
A carbon atom with a larger coefficient will be preferentially attacked, due to more favorable orbital overlap with the electrophile.
[16] The perturbation of a conjugating electron-withdrawing or electron-donating group causes the π electron distribution on a benzene ring to resemble (very slightly!)
The latter species admit tractable quantum calculation using Hückel theory: the cation withdraws electron density at the ortho and para positions, favoring meta attack, whereas the anion releases electron density into the same positions, activating them for attack.
For example, aniline has resonance structures with negative charges around the ring system:Attack occurs at ortho and para positions, because the (partial) formal negative charges at these positions indicate a local electron excess.
Another common argument, which makes identical predictions, considers the stabilization or destabilization by substituents of the Wheland intermediates resulting from electrophilic attack at the ortho/para or meta positions.
Inductively, the negatively charged carboxylate ion moderately repels the electrons in the bond attaching it to the ring.
[10] These groups have a strong electron-withdrawing inductive effect (-I) either by virtue of their positive charge or because of the powerfully electronegativity of the halogens.
Because inductive effects depends strongly on proximity, the meta and ortho positions of fluorobenzene are considerably less reactive than benzene.
However, bromobenzene and iodobenzene are about the same or a little more reactive than chlorobenzene, because although the resonance donation is even worse, the inductive effect is also weakened due to their lower electronegativities.
Thus the overall order of reactivity is U-shaped, with a minimum at chlorobenzene/bromobenzene (relative nitration rates compared to benzene = 1 in parentheses): PhF (0.18) > PhCl (0.064) ~ PhBr (0.060) < PhI (0.12).
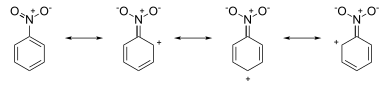
Oppositely, withdrawing electron density is more favourable: (see the picture on the right).As a result, the nitroso group is a deactivator.
However, it has available to donate electron density to the benzene ring during the Wheland intermediate, making it still being an ortho / para director.