Aza-Cope rearrangement
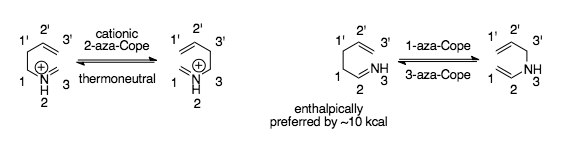
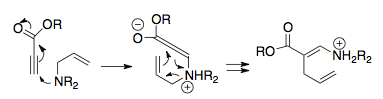
[2] The facile nature of this rearrangement is attributed both to the fact that the cationic 2-aza-Cope is inherently thermoneutral, meaning there's no bias for the starting material or product, as well as to the presence of the charged heteroatom in the molecule, which lowers the activation barrier.
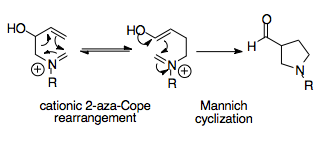
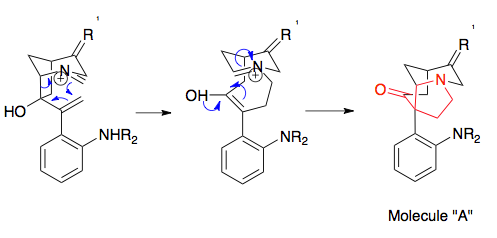
The most common and synthetically useful strategy couples the cationic 2-aza-Cope rearrangement with a Mannich cyclization, and is the subject of much of this article.
It provides easy access to acyl-substituted pyrrolidines, a structure commonly found in natural products such as alkaloids, and has been used in the synthesis of a number of them, notably strychnine and crinine.
It is the most extensively studied of the aza-Cope rearrangements due to the mild conditions required to carry the arrangement out, as well as for its many synthetic applications, notably in alkaloid synthesis.
[1] In 1950, Horowitz and Geissman reported the first example of the 2-aza-Cope rearrangement, a surprising result in a failed attempt to synthesize an amino alcohol.
[9] As is the trend with many reactions, conversion of the Z-enolate affords lower selectivity due to 1,3 diaxial steric interactions between the enolate and the ring, as well as the fact that substituents prefer quasi-equatorial positioning.
[11] Significantly, these stereochemical experiments imply that the cationic 2-aza-Cope rearrangement (as well as Mannich cyclization) occur faster than enol or iminium tautomerization.
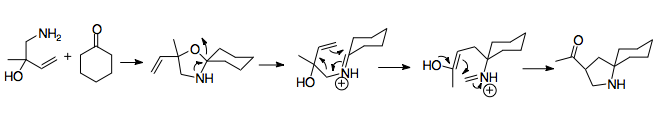
[1] The aza-Cope/Mannich reaction, when participating in ring-expanding annulations, follows the stereochemistry dictated by the most favorable chair conformation, which generally places bulky substituents quasi-equatorially.
[12][13] For simple aza-Cope/Mannich reactions that do not participate in ring-expanding annulation, namely condensations of amino alcohols and ethers, bond rotation occurs more quickly than the Mannich cyclization, and racemic products are observed.
[1] Horowitz and Geissman's first example demonstrates a possible thermodynamic sink to couple with the cationic 2-aza-Cope rearrangement, where the product is biased by the phenyl substituent through aryl conjugation, then captured by hydrolysis of the iminium.
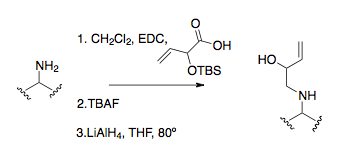
[1][16] Overman and coworkers recognized that the cationic 2-aza-Cope rearrangement could potentially be synthetically powerful if an appropriate thermodynamic sink could be introduced.
Their logic was to incorporate a nucleophilic substituent into the starting material, namely an alcohol group, which acts only after rearrangement, converted into an enol primed to attack the iminium ion.
The aza-Cope/Mannich reaction forces each atom in the [1,5] diene analog to undergo sp2 hybridization, erasing the starting material's stereochemistry at the labelled R' position, while the aza-Prins/pinacol rearrangement retains stereochemistry at the labelled R' position, pointing to a simple test that reveals the active mechanism.
A simple experiment verified that the product was racemic, providing clear evidence of the aza-Cope Mannich reaction as the operative mechanism.
[14] Recent literature from the Shanahan lab supports the rare aza-Prins/pinacol pathway only associated with significantly increased alkene nucleophilicity and iminium electrophilicity.
The stereochemistry of the rearrangement is slightly more complicated when the allyl and amine substituents are installed on a ring, and thus cis or trans to one another.
Strychnine is a naturally occurring highly poisonous alkaloid, found in the tree and climbing shrub genus Strychnos.
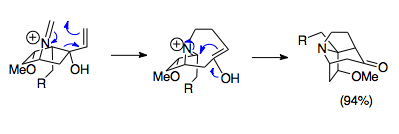
The example shown is a facile reaction combining a 1-aza-bicyclo[2.2.1]heptane salt starting material with paraformaldehyde at 80 °C to form the pivotal aza-tricyclic structure of the Stemona alkaloid molecules.
Seven-membered ring cycles are also possible to synthesize, as the enol and iminium ions stay in close enough proximity to undergo Mannich cyclization.
This rearrangement first creates the vinyl oxazolidine from attack on the cyclohexanone by the aminobutenol, which then undergoes the aza-Cope/Mannich reaction using heat and acid (Lewis or protic).
More complex examples attach the oxazolidine to another ring, presenting additional methods for the formation of indolizidine cycles.
The reaction exhibits high diastereoselectivity, and is robust, proceeding even when faced with poor orbital overlap in the transition state.
[9] Ketones and sterically hindered aldehydes are not suitable for the basic aza-Cope/Mannich reaction, as the amine cannot form an iminium ion with them.
Dehydrative oxazoline formation followed by heating in the presence of a full equivalent of acid present a way to get around this issue.
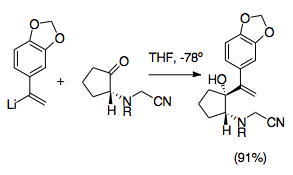
[6][25] Cyanomethyl groups represent an easy way to protect an iminium ion during allylic vinylation of the ketone.
They are typically installed by nucleophilic addition onto an iminium ion, generally produced by amine alkylation with formaldehyde.
The cyanomethyl group protects the nitrogen at the 2-position during formation of the other allylic analog by logic similar to cyanide-type umpolung.
Upon creation of appropriately substituted enamines, intense heating afforded an almost complete rearrangement to the imine product.
This strategy was shown to be relatively robust, allowing for the formation of products even when forced through a boat transition state, when perturbed with substituent effects, or put in competition with alternative rearrangements.
[3] Other methods of overcoming this thermodynamic barrier include pairing it with cyclopropane ring strain release, which allows the reaction to proceed at much lower temperatures.