Carborane acid
Their high acidities stem from the extensive delocalization of their conjugate bases, carboranate anions (CXB11Y5Z6−), which are usually further stabilized by electronegative groups like Cl, F, and CF3.
Due to the lack of oxidizing properties and the exceptionally low nucleophilicity and high stability of their conjugate bases, they are the only superacids known to protonate C60 fullerene without decomposing it.
[5][6] Additionally, they form stable, isolable salts with protonated benzene, C6H7+, the parent compound of the Wheland intermediates encountered in electrophilic aromatic substitution reactions.
[17][16] Carborane acid was first discovered and synthesized by Professor Christopher Reed and his colleagues in 2004 at the University of California, Riverside.
[21] Nevertheless, the synthesis of carborane acids remains lengthy and difficult and requires a well-maintained glovebox and some specialized equipment.
The final step of the synthesis is the metathesis of the μ-hydridodisilylium carboranate salt with excess liquid, anhydrous hydrogen chloride, presumably driven by the formation of strong Si–Cl and H–H bonds in the volatile byproducts: The product was isolated by evaporation of the byproducts and was characterized by its infrared (νCH = 3023 cm−1) and nuclear magnetic resonance (δ 4.55 (s, 1H, CH), 20.4 (s, 1H, H+) in liquid SO2) spectra (note the extremely downfield chemical shift of the acidic proton).
[21] Although the reactions used in the synthesis are analogous, obtaining a pure sample of the more acidic H(CHB11F11) turned out to be even more difficult, requiring extremely rigorous procedures to exclude traces of weakly basic impurities.
The chlorine atoms serve to enhance acidity and act as shields against attacks from the outside due to the steric hindrance they form around the cluster.
[22] This was shown in a single crystal X-ray diffraction study revealing shortened bond lengths in the heterocyclic portion of the ring suggesting electronic delocalization.
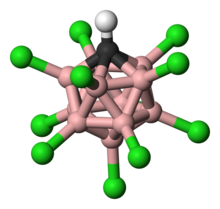
The descriptor closo indicates that the molecule is formally derived (by B-to-C+ replacement) from a borane of stoichiometry and charge [BnHn]2− (n = 12 for known carborane acids).
[24] The cagelike structure formed by the 11 boron atoms and 1 carbon atom allows the electrons to be highly delocalized through the 3D cage (the special stabilization of the carborane system has been termed "σ-aromaticity"), and the high energy required to disrupt the boron cluster portion of the molecule is what gives the anion its remarkable stability.
[27] In inorganic synthesis, their unparalleled acidity may allow for the isolation of exotic species like salts of protonated xenon.


11 Cl
11 ) was shown to be monomeric in the gas phase, with the acidic proton (shown in red ) bound to Cl(12) and secondarily bonded to Cl(7). The monomeric form is metastable when condensed, but eventually polymerizes to give a structure with the acid proton bridging between carborane units. [ 10 ] ( N.B. : The lines between the carbon and boron atoms of the carborane core show connectivity but should not be interpreted to be single bonds. The bond orders are less than one, due to electron deficiency.)


