Catalytic resonance theory
In chemistry, catalytic resonance theory was developed to describe the kinetics of reaction acceleration using dynamic catalyst surfaces.
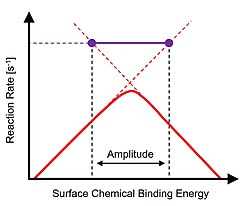
Optimal catalyst performance is depicted as a 'volcano' peak using a descriptor of the chemical reaction defining different catalytic materials.
[3] As described, extension of either side of the volcano plot above the peak defines the timescales of the two rate-limiting phenomena such as surface reaction(s) or desorption.
[8] In the transition between surface binding energy amplitude endpoints, the instantaneous reaction rate (i.e., turnover frequency) oscillates over an order of magnitude as a limit cycle solution.
For any non-unity γi-j system, the asymmetry in the surface energy profile results in conducting work to bias the reaction to a steady state away from equilibrium.
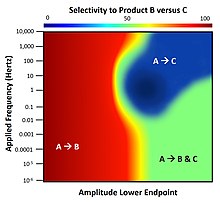
In the range of 1-10 Hertz, there exists a small island of parameters for which product C is highly selective; this condition is only accessible via dynamics.
Perturbation of the active site with strain, light, or condensed charge modulates the binding energy of adsorbates and transition states to alter the rate, selectivity, and conversion of a catalytic reaction.
New opportunities exist with dynamic catalysts and oscillating free energy surfaces not possible with conventional static active sites.
For weaking catalyst binding conditions, B* readily desorbs to form B(g), as A(g) immediately adsorbs as A* to restart the catalytic cycle.
However, in the weak-binding catalyst state, B* readily reacts backwards to reform A* rather than desorbing to form B(g) in the gas phase.
[17] An alternative programmable catalytic mechanism leading to reduced turnover efficiency derives from the extent of the surface that participates in the overall reaction.
The kinetic bias of an independent catalytic ratchet exists for sufficiently high catalyst oscillation frequencies, f, above the ratchet cutoff frequency, fc, calculated as: For a single independent catalytic elementary step of a reaction on a surface (e.g., A* ↔ B*), the A* surface coverage, θA, can be predicted from the ratchet directionality metric, Catalytic rate enhancement via dynamic perturbation of surface active sites has been demonstrated experimentally with dynamic electrocatalysis and dynamic photocatalysis.
Those results may be explained in the framework of catalytic resonance theory but conclusive evidence is still lacking: Implementation of catalyst dynamics has been proposed to occur by additional methods using oscillating light, electric potential, and physical perturbation.