Cell cycle checkpoint
[2] Progression through these checkpoints is largely determined by the activation of cyclin-dependent kinases by regulatory protein subunits called cyclins, different forms of which are produced at each stage of the cell cycle to control the specific events that occur therein.
[8] The main mechanism of action of the cell cycle checkpoints is through the regulation of the activities of a family of protein kinases known as the cyclin-dependent kinases (CDKs), which bind to different classes of regulator proteins known as cyclins, with specific cyclin-CDK complexes being formed and activated at different phases of the cell cycle.
As the cell progresses through G1, depending on internal and external conditions, it can either delay G1, enter a quiescent state known as G0, or proceed past the restriction point.
[10] The three pocket proteins are Retinoblastoma (Rb), p107, and p130, which bind to the E2F transcription factors to prevent progression past the G1 checkpoint.
[10] Positive feedback plays an essential role in regulating the progression from G1 to S phase, particularly involving the phosphorylation of Rb by a Cyclin/CDK protein complex.
During the beginning of the G1 phase, growth factors and DNA damage signal for the rise of cyclin D levels, which then binds to Cdk4 and Cdk6 to form the CyclinD:Cdk4/6 complex.
Each of the fourteen specific mono-phosphorylated isoforms has a differential binding preference to E2F family members, which likely adds to the diversity of cellular processes within the mammalian body.
In many genetic control networks, positive feedback ensures that cells do not slip back and forth between cell cycle phases [12] Cyclin E:Cdk2 proceeds to phosphorylate Rb at all of its phosphorylation sites, also termed “hyper-phosphorylate”, which ensures complete inactivation of Rb.
In turn, this allows for full activation of Cyclin A:Cdk2, a complex which phosphorylates E2F 1-3 initiating their disassociation from the DNA promoter sites.
To maintain the arrest, another response is initiated, by which Chk2 or Chk1 phosphorylate p53, a tumor suppressor, and this stabilizes p53 by preventing it from binding Mdm2, a ubiquitin ligase which inhibits p53 by targeting it for degradation.
The stable p53 then acts a transcriptional activator of several target genes, including p21, an inhibitor of the G1-to-S promoting complex cyclin E-CDK2.
Although variations in requisite cyclin-Cdk complexes exist across organisms, the necessity of the kinase activity is conserved and typically focuses on a single pairing.
As the cell progresses through G2 and reaches the G2/M transition, the kinase Plk1 phosphorylates Wee1, which targets Wee1 for degradation via the SCF ubiquitin ligase complex.
This exists at a level higher than the minimum needed for the continuation of M phase after entry, acting to safeguard the all-or-nothing event.
This entry concentration is further increased in the case of incomplete DNA replication, adding another regulatory mechanism at the G2/M transition point.
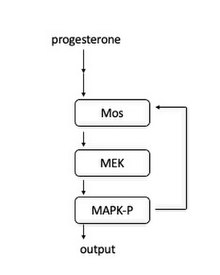
The activation of Mos leads to a positive feedback loop and therefore acts as “toggle switch” to create the all-or-nothing entrance into mitosis.
When the progesterone level is high enough, the Mos curve is shifted higher and ultimately intersects the degradation line at only one point, so there is only one stable “on” state, indicating the entrance into mitosis.
At high enough levels of progesterone, the system is monostable as a result of the positive feedback loop between Mapk and Mos.
So, we can understand the all-or-nothing, irreversible response of the mitotic transition with a mathematical model of the molecular regulators as a bistable system that depends on the existence of positive feedback.
Since entering mitosis is a large and costly commitment for the cell, it is logical that systems would be in place to prevent premature entrance into this step.
It has been shown that mistakes in previous steps, such as having unreplicated sections of DNA blocks progression in the cell cycle.
[27] So, these experiments confirm that the stress of unreplicated DNA in the cell affect the hysteresis loop and result in a much higher cyclin B threshold to enter into mitosis.
To do this, the sensing mechanism ensures that the anaphase-promoting complex (APC/C) is no longer inhibited, which is now free to degrade cyclin B, which harbors a D-box (destruction box), and to break down securin.
[30] Once this inhibitory protein is degraded via ubiquitination and subsequent proteolysis, separase then causes sister chromatid separation.
[32] The loss of ATM has been shown to precede lymphoma development presumably due to excessive homologous recombination, leading to high genomic instability.
[33] Disruption of Chk1 in mice led significant misregulation of cell cycle checkpoints, an accumulation of DNA damage, and an increased incidence of tumorigenesis.
BRCA2 is believed to be involved in homologous recombination and regulating the S-phase checkpoint, and mutations of deficiencies in BRCA2 are strongly linked to tumorigenesis.