Familial hypercholesterolemia
[1] There are five types of familial dyslipidemia (not including subtypes), and each are classified from both the altered lipid profile and by the genetic abnormality.
[2] People who have one abnormal copy (are heterozygous) of the LDLR gene may develop cardiovascular disease prematurely at the age of 30 to 40.
[citation needed] Heterozygous FH is normally treated with statins, bile acid sequestrants, or other lipid-lowering agents that lower cholesterol levels.
[4][5] Accelerated deposition of cholesterol in the walls of arteries leads to atherosclerosis, the underlying cause of cardiovascular disease.
Less commonly, arteries of the brain are affected; this may lead to transient ischemic attacks (brief episodes of weakness on one side of the body or inability to talk) or occasionally stroke.
Peripheral artery occlusive disease (obstruction of the arteries of the legs) occurs mainly in people with FH who smoke; this can cause pain in the calf muscles during walking that resolves with rest (intermittent claudication) and problems due to a decreased blood supply to the feet (such as gangrene).
[6] Atherosclerosis risk is increased further with age and in those who smoke, have diabetes, high blood pressure and a family history of cardiovascular disease.
The related disease sitosterolemia, which has many similarities with FH and also features cholesterol accumulation in tissues, is due to ABCG5 and ABCG8 mutations.
[9] It comprises 18 exons and spans 45 kb, and the protein gene product contains 839 amino acids in mature form.
[4][13] Mutations in the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene were linked to autosomal dominant (i.e. requiring only one abnormal copy) FH in a 2003 report.
In many heterozygous forms of FH, the receptor function is only mildly impaired, and LDL levels will remain relatively low.
[4] Some studies of FH cohorts suggest that additional risk factors are generally at play when a person develops atherosclerosis.
[20] Several studies found that a high level of lipoprotein(a) was an additional risk factor for ischemic heart disease.
[6] A 2007 meta-analysis found that "the proposed strategy of screening children and parents for familial hypercholesterolaemia could have considerable impact in preventing the medical consequences of this disorder in two generations simultaneously.
[34] Prior to the introduction of the statins, clofibrate (an older fibrate that often caused gallstones), probucol (especially in large xanthomas) and thyroxine were used to reduce LDL cholesterol levels.
Rather, evidence of benefit is derived from several trials conducted in people who have polygenic hypercholesterolemia (in which heredity plays a smaller role).
Still, a 1999 observational study of a large British registry showed that mortality in people with FH had started to improve in the early 1990s when statins were introduced.
[37] Alirocumab and evolocumab, both monoclonal antibodies against PCSK9, are specifically indicated as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia, who require additional lowering of LDL cholesterol.
[4] Very severe cases may be considered for a liver transplant; this provides a liver with normally functional LDL receptors, and leads to rapid improvement of the cholesterol levels, but at the risk of complications from any solid organ transplant (such as rejection, infections, or side-effects of the medication required to suppress rejection).
[43][44][45] Lomitapide, an inhibitor of the microsomal triglyceride transfer protein,[46] was approved by the US FDA in December 2012 as an orphan drug for the treatment of homozygous familial hypercholesterolemia.
[47] In January 2013, The US FDA also approved mipomersen, which inhibits the action of the gene apolipoprotein B, for the treatment of homozygous familial hypercholesterolemia.
[52] Given that FH is present from birth and atherosclerotic changes may begin early in life,[53] it is sometimes necessary to treat adolescents or even teenagers with agents that were originally developed for adults.
Due to safety concerns, many physicians prefer to use bile acid sequestrants and fenofibrate as these are licensed for children.
[6][54] An expert panel in 2006 advised early combination therapy with LDL apheresis, statins, and cholesterol absorption inhibitors in children with homozygous FH at the highest risk.
The Afrikaner, French Canadians, Lebanese Christians, and Finns have high rates of specific mutations that make FH particularly common in these groups.
[4] The Norwegian physician Dr Carl Müller first associated the physical signs, high cholesterol levels, and autosomal dominant inheritance in 1938.
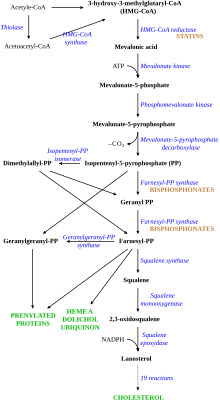
Initially, they found increased activity of HMG-CoA reductase, but studies showed that this did not explain the very abnormal cholesterol levels in people with FH.
Subsequent selection and breeding produced forms with heightened susceptibility of coronary atherosclerosis and myocardial infarction, as hypercholesterolemia alone in rabbits was not sufficient to cause this issues frequently enough.