Hiyama coupling
This reaction was discovered in 1988 by Tamejiro Hiyama and Yasuo Hatanaka as a method to form carbon-carbon bonds synthetically with chemo- and regioselectivity.
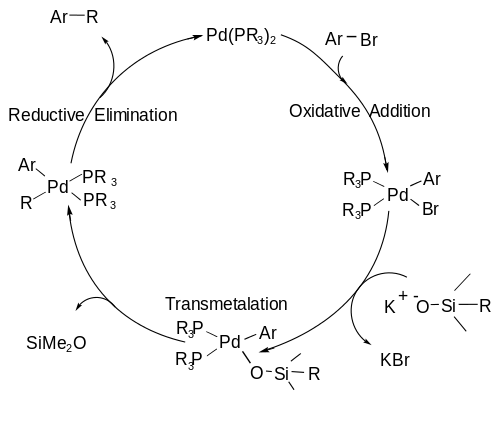
The reaction first reported was used to couple easily made (and activated) organosilicon nucleophiles and organohalides (electrophiles) in the presence of a palladium catalyst.
[1] Since this discovery, work has been done by various groups to expand the scope of this reaction and to "fix" the issues with this first coupling, such as the need for fluoride activation of the organosilane.
The organosilane is activated with fluoride (as some sort of salt such as TBAF or TASF) or a base to form a pentavalent silicon center which is labile enough to allow for the breaking of a C-Si bond during the transmetalation step.
[9] A nickel catalyst allows for access to new reactivity of organotrifluorosilanes as reported by GC Fu et al.[10] Secondary alkyl halides are coupled with aryl silanes[11] with good yields using this reaction.
Addition of fluoride cleaves any silicon protecting groups (e.g. silyl ethers[12]), which are frequently employed in organic synthesis.
To overcome this issue, many groups have looked to the use of other basic additives for activation, or use of a different organosilane reagent all together, leading to the multiple variations of the original Hiyama coupling.
The general reaction scheme is shown below, showcasing the utilization of a Brønsted base as the activating agent as opposed to fluoride, phosphine ligands are also used on the metal center.
The presence of a pentavalent silicon is not needed and kinetic analysis has shown that this reaction has first order dependence on silonate concentration.