Iron–sulfur protein
Fe–S proteins are vulnerable to attack by biogenic nitric oxide, forming dinitrosyl iron complexes.
[3] This high covalency lowers the inner sphere reorganization energy[3] and ultimately contributes to a rapid electron transfer.
The Rieske proteins contain Fe–S clusters that coordinate as a 2Fe–2S structure and can be found in the membrane bound cytochrome bc1 complex III in the mitochondria of eukaryotes and bacteria.
These photosynthetic organisms include plants, green algae, and cyanobacteria, the bacterial precursor to chloroplasts.
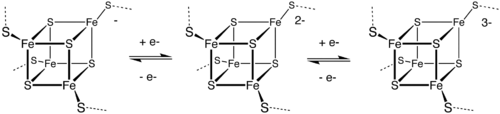
Low- and high-potential ferredoxins are related by the following redox scheme: In HiPIP, the cluster shuttles between [2Fe3+, 2Fe2+] (Fe4S42+) and [3Fe3+, Fe2+] (Fe4S43+).
The potentials for this redox couple range from 0.4 to 0.1 V. In the bacterial ferredoxins, the pair of oxidation states are [Fe3+, 3Fe2+] (Fe4S4+) and [2Fe3+, 2Fe2+] (Fe4S42+).
The potentials for this redox couple range from −0.3 to −0.7 V. The two families of 4Fe–4S clusters share the Fe4S42+ oxidation state.
The difference in the redox couples is attributed to the degree of hydrogen bonding, which strongly modifies the basicity of the cysteinyl thiolate ligands.
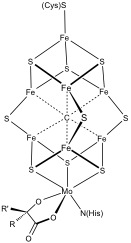
[citation needed] A further redox couple, which is still more reducing than the bacterial ferredoxins is implicated in the nitrogenase.
In aconitase, the Fe–S cluster binds aconitate at the one Fe centre that lacks a thiolate ligand.
The cluster does not undergo redox, but serves as a Lewis acid catalyst to convert citrate to isocitrate.
Examples include the active sites of a number of enzymes: The biosynthesis of the Fe–S clusters has been well studied.
[15][16][17] The biogenesis of iron sulfur clusters has been studied most extensively in the bacteria E. coli and A. vinelandii and yeast S. cerevisiae.