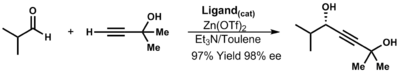Organozinc chemistry
In 1848 Edward Frankland prepared the first organozinc compound, diethylzinc, by heating ethyl iodide in the presence of zinc metal.
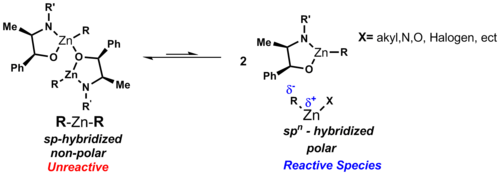
The dipole moment of symmetric diorganozinc reagents can be seen as zero in these linear complexes, which explains their solubility in nonpolar solvents like cyclohexane.
Unlike other binary metal alkyls, the diorganozinc species show a low affinity for complexation with ethereal solvent.
Therefore, it is rare for bridging alkyl or aryl groups to occur due to the weak electron deficiency of the zinc atom.
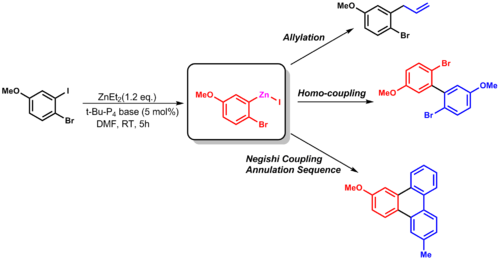
In one study[8][9] the active organozinc compound is obtained from much cheaper organobromine precursors: Frankland's original synthesis of diethylzinc involves the reaction of ethyl iodide with zinc metal.
Formation of organozinc reagents is facilitated for alkyl or aryl halides bearing electron-withdrawing substituents, e.g., nitriles and esters.
[10][11] The two most common zinc functional group interconversion reactions are with halides and boron, which is catalyzed by copper iodide (CuI) or base.
This synthesis shows the utility of organozinc reagents by displaying high selectivity for the most reactive site in the molecule, as well as creating useful coupling partners.
The initial step is an oxidative addition of zinc metal into the carbon-halogen bond, thus forming a carbon-zinc enolate.
The key zinc-intermediate formed is a carbenoid (iodomethyl)zinc iodide which reacts with alkenes to afford the cyclopropanated product.
The rate of forming the active zinc species is increased via ultrasonication since the initial reaction occurs at the surface of the metal.
[21] Organozinc compounds derived from methylene bromide or iodide can electrophilically add to carbonyl groups to form terminal alkenes.
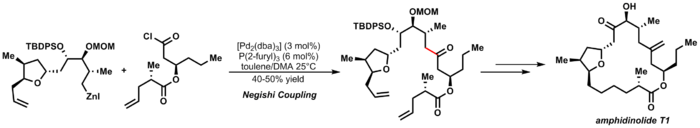
[26] Either diorganic[check spelling] species or organozinc halides can be used as coupling partners during the transmetallation step in this reaction.
A key step in the catalytic cycle is a transmetalation in which a zinc halide exchanges its organic substituent for another halogen with the metal center.
This thioester compound can be coupled to a wide range of organozinc reagents in order to reveal the corresponding ketone product.
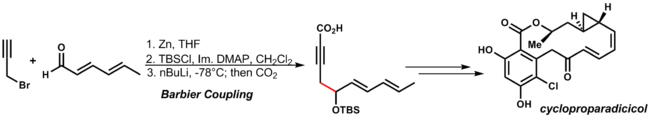
In this case, the Fukuyama coupling takes place with the thiolactone:[31] The Barbier reaction involves nucleophilic addition of a carbanion equivalent to a carbonyl.
The Barbier reaction is advantageous because it is a one-pot process: the organozinc reagent is generated in the presence of the carbonyl substrate.
[35] Diastereoselectivity for addition of organozinc reagents into aldehydes can be predicted by the following model by Noyori and David A. Evans:[36] Zinc-acetylides are used in the HIV-1 reverse transcriptase inhibitor Efavirenz as well as in Merck's ephedrine derivatives .
One example is [(Me3SiCH2)3Zn]K. Triethylzincate degrades to sodium hydridoethylzincate(II) as a result of beta-hydride elimination:[39] The product is an edge-shared bitetrahedral structure, with bridging hydride ligands.
Although less commonly studied, organozincates often have increased reactivity and selectivity compared to the neutral diorganozinc compounds.