Sharpless asymmetric dihydroxylation
The reaction has been applied to alkenes of virtually every substitution, often high enantioselectivities are realized, with the chiral outcome controlled by the choice of dihydroquinidine (DHQD) vs dihydroquinine (DHQ) as the ligand.
This reaction was developed principally by K. Barry Sharpless building on the already known racemic Upjohn dihydroxylation, for which he was awarded a share of the 2001 Nobel Prize in Chemistry.
Methanesulfonamide (CH3SO2NH2) has been identified as a catalyst to accelerate this step of the catalytic cycle and if frequently used as an additive to allow non-terminal alkene substrates to react efficiently at 0 °C.
[20] In the February 1997 issue of the Journal of the American Chemical Society Sharpless published the results of a study (a Hammett analysis) which he claimed supported a [2+2] cyclization over a [3+2].
[21] In the October issue of the same year, however, Sharpless also published the results of another study conducted in collaboration with Ken Houk and Singleton which provided conclusive evidence for the [3+2] mechanism.
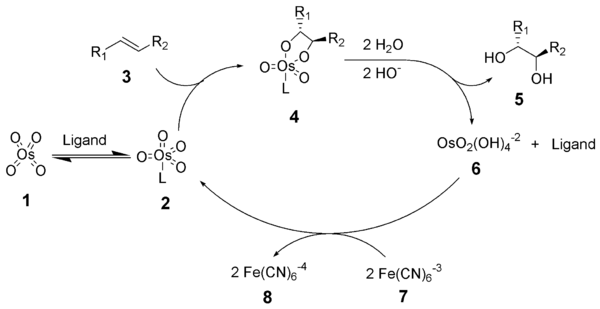
Crystallographic evidence has shown that the active catalyst possesses a pentacoordinate osmium species held in a U-shaped binding pocket.
Given below is a brief overview of the various components of the catalytic system: In general Sharpless asymmetric dihydroxylation favors oxidation of the more electron-rich alkene (scheme 1).
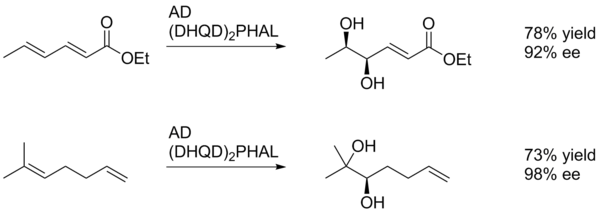
[23] The diastereoselectivity of SAD is set primarily by the choice of ligand (i.e. AD-mix-α versus AD-mix-β), however factors such as pre-existing chirality in the substrate or neighboring functional groups may also play a role.
In the example shown below, the para-methoxybenzoyl substituent serves primarily as a source of steric bulk to allow the catalyst to differentiate the two faces of the alkene.
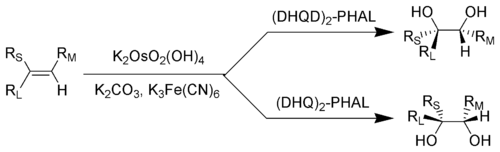
R L = Largest substituent; R M = Medium-sized substituent; R S = Smallest substituent