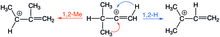
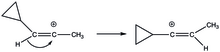Vinyl cation
[3] Vinyl cations have long been poorly-understood[4] and were initially thought to be too high energy to form as reactive intermediates.
Vinyl cations were first proposed in 1944 as a reactive intermediate for the acid-catalyzed hydrolysis of alkoxyacetylenes to give alkyl acetate.
Noyce and coworkers, for example, reported the formation of a vinyl cation in acid-catalyzed hydration of phenylpropiolic acid.
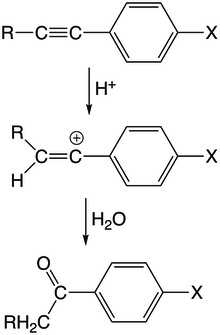
[8] The authors note that in the rate limiting step, a large positive charge develops on the benzylic carbon, indicating that the reaction proceeds through a vinyl cation transition state.
[4] The introduction of “super” leaving group in the 1970s first allowed for the generation of vinyl cation reactive intermediates with appreciable lifetimes.
Utilization of these super leaving groups allowed researchers for the first time to move beyond speculation about the existence of such vinyl cations.
Hinkle and coworkers synthesized a number of alkenyl(aryl)iodonium triflates from hypervalent phenyliodo precursors.
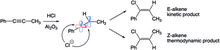
The presence of both E- and Z-vinyl triflate products offers support for the formation of a primary vinyl cation reactive intermediate; through SN2 chemistry, both only one isomer would form.
The reactive intermediate is prone to either nucleophilic attack by the solvent to yield E- and Z-enol ether isomers, or beta hydrogen elimination.
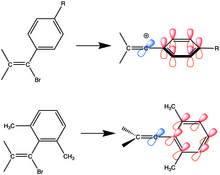
Adding steric bulk to the ortho-positions improve conjugation as it makes the phenyl ring orthogonal to the vinyl carbons but coplanar with the empty p-orbital.
When it is in its bisected structure, there is suitable overlap between its empty p-orbital and the cyclopropyl ring that stabilization is achieved.
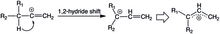
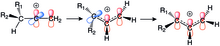
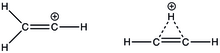
The stabilizing power of the cyclopropyl ring is so great that it has become a driving thermodynamic force in rearrangements like 1,2-hydride shifts in (E)- and (Z)-3-cyclopropyl-2-propenyl triflate solvolysis.
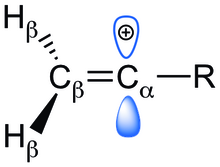
[20] Substituent effects on vinyl cation stability Table 1: Electronic effects responsible for stabilization of vinyl cation at the α-position ^ ‘-’ electron-withdrawing, ‘+’ electron-donating # ‘+’ indicates stabilization and ‘-‘ indicates destabilization of the substituted vinyl cation with respect to neutral alkene equivalent *indicates the strongest factor responsible for (de)stabilization for substituents that exhibit more than one electronic effect ** the substituent is inductively withdrawing at the carbonyl carbon and also exhibits small electron delocalization from the carbonyl oxygen *** Y = -F, -Cl, -Br, -I, -OH, -CN, -CF3 The presence of an empty p-orbital perpendicular to the p-bond imparts unwanted destabilization onto the vinyl cation.
In a preliminary work, 4 substituents (-CH=CH2, -F, -Cl, -CH3) were initially studied to investigate electronic effects on vinyl cation stability.
On the other hand, the increase in the Cb-H bond length implies a strong hyperconjugative effect that is inversely related to the thermodynamic stability of the cation.
Enthalpy calculations obtained from the isodesmic reaction are fair accurate and shows good correlation with experimental data.
Heteroatoms like fluorine and chlorine, can exhibit both inductive (electron-withdrawing) and p-donation electronic effects because of their high electronegativities and p-electrons.
In the acid-catalyzed hydration of arylacetylene derivatives, a proton initially attacks the triple bond to form a vinyl cation at the aryl substituted carbon.
The intermediate experiences little resonance stabilization because of the orthogonality of the conjugated aryl orbital with the empty p-orbital of the vinyl cation.
Another possible explanation is that smaller size of the hydrogen substituent allows solvation to take place more easily contributing more significant stabilization.
The reaction with hydrogen halides, which also has an initial protonation step, results in the formation of halo-substituted alkenes.
Though the E-alkene is initially formed, it isomerizes to the Z-alkene through a carbocation intermediate the stems from protonation and C-C rotation steps.
An alkyne that is adjacent to a tertiary alcohol forms a four-membered cyclic vinyl cation intermediate in which the oxygen of the hydroxyl group bridges two carbons across two bonds.
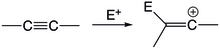
[24] In the electrophilic attack of allenes, it takes place in a manner that prefers to form a terminal adduct and the vinyl cation at the central carbon.
The polarization of the allene group show that the terminal carbons have a higher electron density and tendency to under nucleophilic attack.
However, if the terminal end is stabilized by a substituent, an allyl-like cation may form as the electrophile attacks the central carbon.
Similar to phenyl rings adjacent to vinyl cations, there must be bond rotation to achieve complete resonance stabilization.
In the reaction of 5-chloropent-1-yne with trifluoroacetic acid, there is simultaneous protonation and 1,4-shift of chlorine that forms a bridged cyclic structure across four carbons.
[18] Ketenes and allenes undergo [2+2] cycloadditions under thermal conditions in a concerted manner because they have pi orbitals that are orthogonal to each other.
[26] There is debate on whether a vinyl cation intermediate forms with the addition of a halide (H-X) compound to a terminal alkyne for hydrohalogenation reactions.



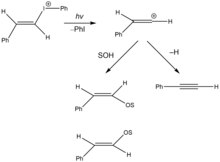
2 H +
3 . Adapted from [ 17 ]