Discovery and development of direct Xa inhibitors
Heparin was discovered by Jay McLean and William Henry Howell in 1916, it was first isolated from a canine liver, which in Greek translates to hepar.
[6] Warfarin treatment requires blood monitoring and dose adjustments regularly due to its narrow therapeutic window.
If supervision isn't adequate warfarin poses a threat in causing, all too frequent, haemorrhagic events and multiple interactions with food and other drugs.
[8] Direct Xa inhibitors are just as efficacious as LMWH and warfarin but they are given orally and don't need as strict monitoring.
In 1987 the first factor Xa inhibitor, the naturally occurring compound antistasin, was isolated from the salivary glands of the Mexican leech Haementeria officinalis.
[12] After activating various proenzymes, thrombin is formed in the last steps of the cascade, it then converts fibrinogen to fibrin which leads to clot formation.
[10] Factor Xa is an activated serine protease that occupies a key role in the blood coagulation pathway by converting prothrombin to thrombin.
It is a single-chain, 60 amino acid peptide and like antistasin it is a slow, tight-binding inhibitor with a similar Ki value (~0.6 nM).
[8] Those early developed small molecules yet had amidine-groups or even higher-basic functions, which were thought to be necessary as mimics for an arginine residue in prothrombin, the natural substrate of factor Xa.
In 1998 Bayer Healthcare, a pharmaceutical company started searching for low-molecular-weight direct factor Xa inhibitors with higher oral bioavailability.
[8] One compound of this class, Rivaroxaban (IC50 = 0.7 nM, bioavailability: 60%), was granted marketing authorization for the prevention of venous thromboembolism in Europe and Canada in September 2008.
[1][17] Factors IIa, Xa, VIIa, IXa and XIa are all proteolytic enzymes that have a specific role in the coagulation cascade.
[18] The active site of FXa is structured to catalyze the cleavage of physiological substrates and cleaves PhePheAsnProArg-ThrPhe and TyrIleAspGlyArg-IleVal in prothrombin.
It was first noted that the natural compounds, antistasin and TAP, which possess highly polar and therefore charged components bind to the target with some specificity.
Nowadays marketed Xa inhibitors, therefore contain an aromatic ring with various moieties attached for different interactions with the S1 and S4 binding sites.
The Xa inhibitors currently on market today, therefore rely on hydrophobic and hydrogen bonding instead of highly polar interactions.
While FXa has a glutamine residue in that position, thrombin has a glutamic acid that causes electrostatic repulsion with the carboxyl group of DX-9065a.
In addition, a salt bridge between Glu-97 of thrombin and the amidine group fixed in the pyrrolidine ring of DX-9065a reduces the flexibility of the DX-9065a molecule, which now cannot rotate enough to avoid the electrostatic clash.
The interaction between the chlorine substituent of the thiophene moiety and the aromatic ring of Tyr-228, which is located at the bottom of the S1, it is very important due to the fact that it obviates the need for strongly basic groups for high affinity for FXa.
[8]Apixaban shows a similar binding mode as rivaroxaban and forms a tight inhibitor-enzyme complex when connected to FXa.
Modelling both prothrombin and FXa makes it possible to deduct the difference and identify the amino acids at each binding site.
The interaction between the S1 pocket of FXa and the inhibitor can be both ionic or non-ionic, which is important because it allows the design of the moiety to be adjusted to increase oral bioavailability.
Previously designed compounds were charged molecules that are not absorbed well in the gastrointestinal tract and therefore did not reach high serum concentrations.
This compound had a promising pharmacokinetical profile and did not contain a highly basic amidine group, but that had previously been considered important for the interaction with the S1 pocket.
[25] In dog testing, this compound with a carboxamide group called 13F, showed a great pharmacokinetical profile, a low clearance and adequate half-life and volume of distribution.
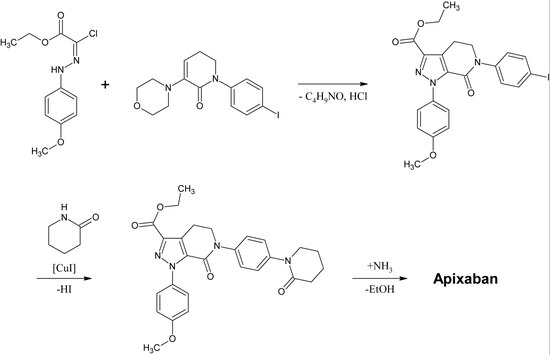
In a one-pot reaction the carbamate cyclisizes to a 2-oxazolidone ring under slightly basic conditions while simultaneously the oxazolidone nitrogen is arylized by copper-catalization.
[27] It starts from N-(4-aminophenol)-morpholinone which is alkylated by a propylene oxide derivate that also contains a primary amine involved in a phthalimide protection group.
The patent also explains another synthesis starting from a chlorothiophene derivate that would be more suitable for the industrial process but points out that toxic solvents or reagents have to be removed from the final product.
After the following elimination of HCl and morpholine, the iodine is substituted by 2-piperidinone by copper-catalization and the ethyl esther is converted to an amide (aminolysis).
[8] Indication for Xa inhibitors is preventing deep vein thrombosis (DVT) which can lead to pulmonary embolism.











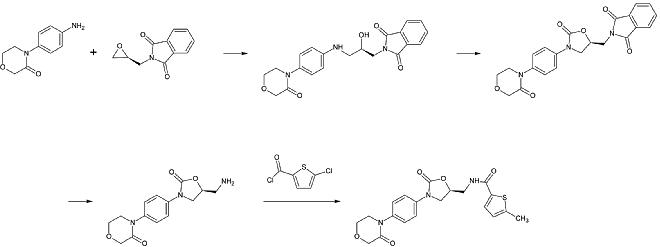
1st step: Alkylation of the primary aromatic amine
2nd step: Formation of the 2-oxazolinidone ring, using a phosgene equivalent
3rd step: Removal of the phthalimide protection group
4th step: Acylation of the primary amine