Discovery and development of proton pump inhibitors
Evidence emerged by the end of the 1970s that the newly discovered proton pump (H+/K+ ATPase) in the secretory membrane of the parietal cell was the final step in acid secretion.
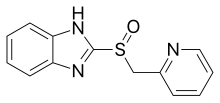
[2][3][4] Timoprazole is a pyridylmethylsulfinyl benzimidazole and appealed due to its simple chemical structure and its surprisingly high level of anti-secretory activity.
[5] Optimization of substituted benzimidazoles and their antisecretory effects were studied on the newly discovered proton pump to obtain higher pKa values of the pyridine, thereby facilitating accumulation within the parietal cell and increasing the rate of acid-mediated conversion to the active mediate.
Hydrolysis of one ATP molecule is used to catalyse the electroneutral exchange of two luminal potassium ions for two cytoplasmic protons through the gastric membrane.
The extracellular domain of the β subunit contains six or seven N-linked glycosylation sites which is important for the enzyme assembly, maturation and sorting.
[9] The expulsion of the proton at 160 mM (pH 0.8) concentration results from movement of lysine 791 into the ion binding site in the E2P configuration.
[6] A derivative of timoprazole, omeprazole, was discovered in 1979, and was the first of a new class of drug that control acid secretion in the stomach, a proton pump inhibitor (PPI).
[6] A new approach for the treatment of acid-related diseases was introduced, and omeprazole was quickly shown to be clinically superior to the histamine H2 receptor antagonists, and was launched in 1988 as Losec in Europe, and in 1990 as Prilosec in the United States.
During the 1980s, about 40 other companies entered the PPIs area, but few achieved market success: Takeda with lansoprazole, Byk Gulden (now Nycomed) with pantoprazole, and Eisai with rabeprazole, all of which were analogues of omeprazole.
Smith Kline and French, that entered into collaboration with Byk Gulden mid-1984, greatly assisted in determining criteria for further development.
[5] Pantoprazole obtained high selection criteria in its development process — especially concerning the favorable low potential for interaction with other drugs.
Good solubility of pantoprazole and a very high solution stability allowed it to become the first marketed PPI for intravenous use in critical care patients.
[5] Omeprazole showed an inter-individual variability and therefore a significant number of patients with acid-related disorders required higher or multiple doses to achieve symptom relief and healing.
Esomeprazole magnesium (brand name Nexium) received its first approval in 2000 and provided more pronounced inhibition of acid secretion and less inter-patient variation compared to omeprazole.
Oral esomeprazole preparations are enteric-coated, due to the rapid degradation of the drug in the acidic condition of the stomach.
[16] Tenatoprazole (TU-199), an imidazopyridine proton pump inhibitor, is a novel compound that has been designed as a new chemical entity with a substantially prolonged plasma half-life (7 hours), but otherwise has similar activity as other PPIs.
[18][21][22][23] Reaction with cysteine 822 confers a rather special property to the covalently inhibited enzyme, namely irreversibility to reducing agents.
Recent data suggest the hydrated sulfenic acid to be the reactive species forming directly from the mono-protonated benzimidazole bound on the surface of the pump.
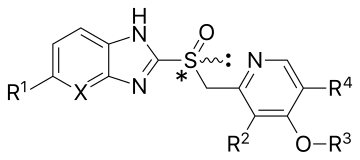
[8] Although the drugs omeprazole, lansoprazole, pantoprazole, and rabeprazole share common structure and mode of action, each differs somewhat in its clinical pharmacology.
Direct comparison of pantoprazole sodium with other anti-secretory drugs showed that it was significantly more effective than H2-receptor antagonists and either equivalent or better than other clinically used PPIs.
[5] Another study states rabeprazole undergoes activation over a greater pH range than omeprazole, lansoprazole, and pantoprazole, and converts to the sulphenamide form more rapidly than any of these three drugs.
[23] Most oral PPI preparations are enteric-coated, due to the rapid degradation of the drugs in the acidic conditions of the stomach.
[30] In Asian as well as Caucasian healthy subjects, tenatoprazole exhibited a seven-fold longer half-life than the existing H+/K+ ATPase inhibitors.
A strong relationship has been stated between the degree and duration of gastric acid inhibition, as measured by monitoring of the 24-hour intragastric pH in pharmacodynamic studies, and the rate of healing and symptom relief reported.
Since the binding is competitive and reversible these agents have the potential to achieve faster inhibition of acid secretion and longer duration of action compared to PPIs, resulting in quicker symptom relief and healing.
[34][36] In June 2006, Yuhan obtained approval from the Korean FDA for the use of revaprazan (brand name Revanex) in the treatment of gastritis.
[37] Vonoprazan is a newer agent with a faster and longer lasting action, first marketed in Japan, then in Russia, and in 2023 was approved for use in the US.